...
UGENE provides the "PCR Primer Design for DNA Assembly" feature only for nucleic sequences with the "Standard DNA" alphabet. To use it in UGENE open a DNA sequence and go to the "PCR Primer Design for DNA Assembly" tab of the Options Panel:
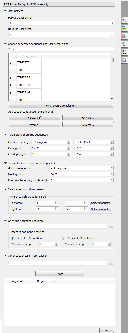

There are the following parameters:
Forward primer - forward primer.
Reverse primer - on the opposite strand from the forward primer.
Mismatches - mismatches limit.
3' perfect match - specify the number of nucleotides at the 3' end that must not have mismatches.
Maximum product - maximum size of the amplified sequence.
Extract annotations - specify the type of extracted annotations: Inner, All intersected or None.
- Value Inner is selected by default. When this value is selected, the extracted PCR product contains annotations from the original sequence, located within the extracted region.
- Value All intersected specifies that all annotations of the original sequence that intersect the extracted region must be extracted as well.
- Value None specifies that annotations from the original sequence must not be extracted.
Choosing primers
Type two primers for running In Silico PCR. If the primers pair is invalid for running the PCR process then the warning is shown. Also, primers for the running In silico PCR can be chosen from a primer library. Click the following button to choose a primer from the primers library:
| HTML |
|---|
<center>
<br>
<img src="/wiki/download/attachments/13435073/Primers Library.png"/>
</br>
</center> |
The following dialog will appear:
| HTML |
|---|
<center>
<br>
<img src="/wiki/download/attachments/16125631/In Silico PCR_2.png"/>
</br>
</center> |
The table consists of the following columns: name, GC-content (%), Tm, Length (bp) and sequence. Select primer in the table and click the Choose button.
Click the Reverse-complement button for making a primer sequence reverse-complement:
| HTML |
|---|
<center>
<br>
<img src="/wiki/download/attachments/13435069/In Silico PCR_1.png"/>
</br>
</center> |
Click Show primers details for seeing statistic details about primers.
When you run the process, the predicted PCR products appear in the products table.
Products table
There are three columns in the table:
- region of product in the sequence
- product length
- preferred annealing temperature
Click the product for navigating to its region in the sequence.
Click the Extract product(s) button for exporting a product(s) in a file or use double click for that.
| HTML |
|---|
<center>
<br>
<img src="/wiki/download/attachments/13435069/In Silico PCR_2.png"/>
</br>
</center> |
...
- User primers - the pair of forward and reverse primers user can check on the absence of unwanted connections (like hairpins, self- and hetero-dimers).
- Choose generated sequences as user primer's end - user primers could be extended with the 8-bases sequences on 5' and 3' ends. These sequences do not form too simple unwanted connections (for example, "AAAATTTT", obviously, has a hairpin, so there is no such a sequence in the list).
- Parameters of priming sequences - the range of melting temperature, Gibbs free energy and length primers SHOULD have.
- Parameters to exclude in whole primers - the minimum melting temperature, the maximum Gibbs free energy and the maximum base pairs length which may be present in the self- and hetero-dimers in the considered primer.
- Select areas for priming search - select areas to search forward and reverse primers in.
- Open the backbone sequence - choose a sequence to be added to 5' or 3' end of the result primer.
- Other sequences in PCR reaction - the list of sequence to check the unwanted connections with. The unwanted connections should fit to "Parameters to exclude in whole primers".
Designing primers
Physical quantity
The designed primers should have the set Gibbs free energy, melting temperature and length. Also, these primers could have hairpins, self- and hetero-dimers, but the mentioned parameters of these found dimers should be limited. Look at the pictures:


The found primer "GATGGTGATGTTAATGGGCAC" has Gibbs free energy between -40 and -30 kcal/mol, melting temperature between 51 and 65 °C and length between 18 and 25 nucleotides. Also, there are no heirpins, self- and hetero-dimers in this primer, which has Gibbs free energy more then -7 kcal/mol, melting temperature less than 20 °C and base pairs length more than 3 nucleotides.
Regions
Forward primers are searched at the left region, reverse primers are searched at the right region. There is an amplified fragment between them:

The left search area is from 1 to 71 base, the right search area is from 355 to 426 base. The amplified fragment between them.
Backbone sequence
Backbone sequence is the sequence that is added to 5' end of the primer for the following assembly of the PCR product with the backbone molecule. This parameter is optional. You may set the backbone sequence, the sequence end you want to add backbone to and the length of the sequence from 5' and 3' ends to check if these sequences have unwanted hairpins, self- and hetero-dimers. On the pictures below you may see the differences between "5' insert to 5' backbone" and "5' insert to 3' backbone" ("insert" in this case is the same as "primer").


If you set "5' backbone length", then the number of bases you set from 5' end of the backbone sequence will be checked for unwanted connections. The same about "3' backbone length", but from the 3' end. If the checked end has hairpins, self- and homo-dimers you will see the following dialog:
| Children Display | ||
|---|---|---|
|
Other sequences in PCR reactions
Sequences that are supposed to be in the reaction mixture. Its important primers do not have unwanted connections to these sequences, so it will be checked that there are no hairpins, self- or hetero-dimers between the resulting primer and any sequence from the "Other sequences in PCR reaction" file.
Result
The search result is from 1 to 4 pairs of primers. The results should be as close as possible to the amplified fragment. That means, that the searching process starts from the edge of the amplified fragment to the 5' direction on the direct strand in case of the forward primer and to the 3' direction of the reverse-complementary strand in case of the reverse primer. The results are defined with special names:
- A - the pair of primers closest to the amplified fragment, corresponding to the entire set of parameters.
- B1 - the pair of primers closest to the amplified fragment, corresponding to the entire set of parameters if B2 and/or B3 exist.
- B2 - the pair of primers corresponding to the entire set of parameters, which has a 4-nucleotide intersection with B1.
- B3 - the pair of primers corresponding to the entire set of parameters, which starts immediately when B1 ends.

Also, you may see results in the result table. A single click on the result in the result table selects the corresponding annotation, double click - export the resulting primer to the separate file (with the backbone sequence, which will be added during the exporting process).
User primer
You may check the validity of the pair of primers by typing this pair into the corresponding fields. Also, you may add your primers 5' or/and 3' 8 bases endings, which do not form simple unwanted connections.