Use this workflow sample to assemble genomes from short reads with Spades.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Assembly with Spades" can be found in the "NGS" section of the Workflow Designer samples.
Workflow Image
There are two versions of the workflow available. The workflow for single tags looks as follows:
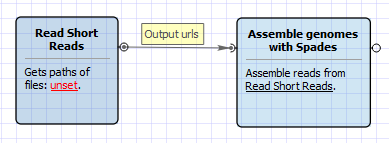
The workflow for paired tags appearance is the following:
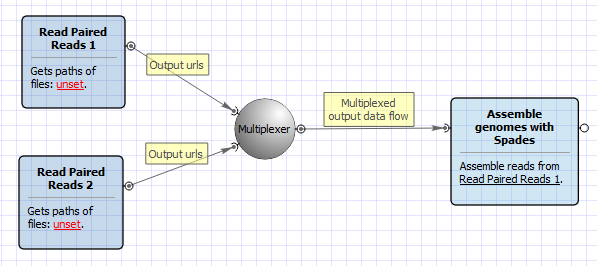
Workflow Wizard
The wizard for single tags has 1 page.
Genome assembly settings: On this page you must input single reads and optionally modify advanced parameters.
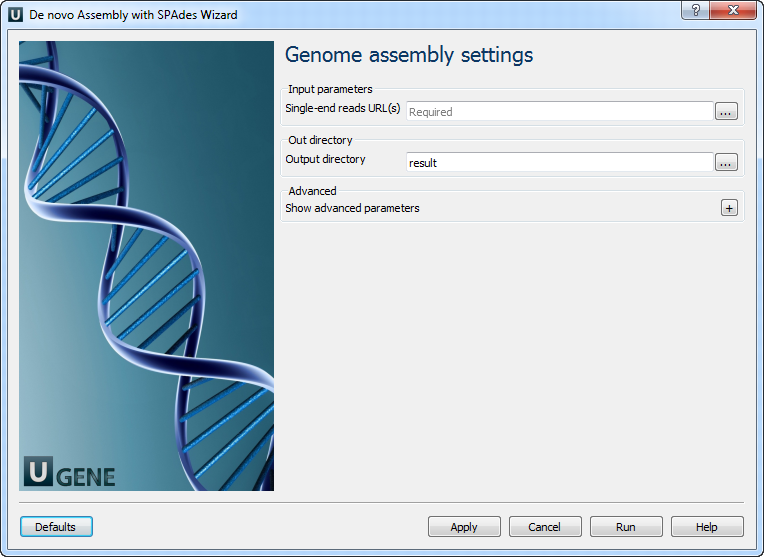
The following parameters are available:
Single-end reads URL(s) Semicolon-separated list of pathes to the input files. Output directory Directory to save Spades output files. Dataset type Input dataset type. Running mode
Running mode.
K-mers
k-mer sizes (-k).
The wizard for paired tags has 1 page.
Genome assembly settings: On this page you must input paired reads and optionally modify advanced parameters.
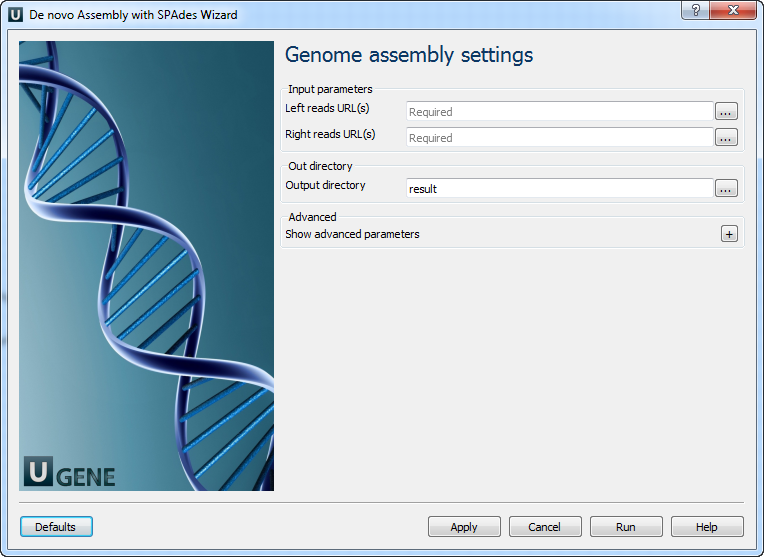
The following parameters are available:
Left reads URL(s) Semicolon-separated list of pathes to the input files. Right reads URL(s) Semicolon-separated list of pathes to the input files. Output directory Directory to save Spades output files. Dataset type Input dataset type. Running mode
Running mode.
K-mers
k-mer sizes (-k).