The workflow sample, described below, prepare ChIP-Seq processed data (with BedTools and bedGraphToBigWig) for visualization in a genome browser. For input BED-file produces BigWig file.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "ChIP-Seq Coverage" can be found in the "NGS" section of the Workflow Designer samples.
Workflow Image
The opened workflow looks as follows:
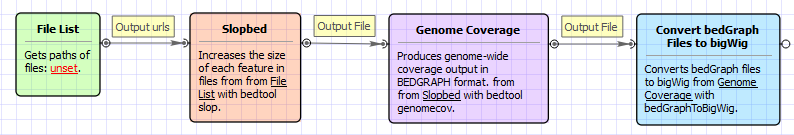
Workflow Wizard
The wizard has 3 pages.
Input data Page: On this page you must input BED file with ChIP-Seq tags.
Parameters Page: Here you can optionally modify parameters that should be used for the Slopbed, Genome Coverage and BedGraphToBigWig elements.
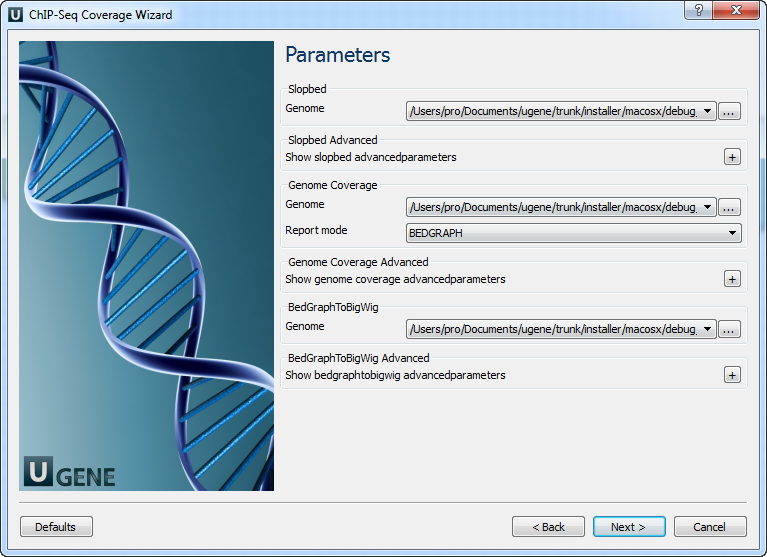
The following parameters are available:
Genome In order to prevent the extension of intervals beyond chromosome boundaries, bedtools slop requires a genome file defining the length of each chromosome or contig. The format of the file is: (-g). Each direction increase Increase the BED/GFF/VCF entry by the same number base pairs in each direction. If this parameter is used -l and -l are ignored. Enter 0 to disable. (-b) Substract from start The number of base pairs to subtract from the start coordinate. Enter 0 to disable. (-l) Add to end
The number of base pairs to add to the end coordinate. Enter 0 to disable. (-r)
Strand-based
Define -l and -r based on strand. For example. if used, -l 500 for a negative-stranded feature, it will add 500 bp to the end coordinate. (-s)
As fraction
Define -l and -r as a fraction of the feature's length. E.g. if used on a 1000bp feature, -l 0.50, will add 500 bp upstream. (-pct)
Print header
Print the header from the input file prior to results. (-header)
Filter start>end fields
Remove lines with start postion greater than end position Report mode Histogram () - Compute a histogram of coverage. Per-base (0-based) (-dz) - Compute the depth of feature coverage for each base on each chromosome (0-based). Per-base (1-based) (-d) - Compute the depth of feature coverage for each base on each chromosome (1-based). BEDGRAPH (-bg) - Produces genome-wide coverage output in BEDGRAPH format. BEDGRAPH (including uncoveded) (-bga) - Produces genome-wide coverage output in BEDGRAPH format (including uncovered). Split Treat BAM or BED12 entries as distinct BED intervals when computing coverage. For BAM files, this uses the CIGAR and operations to infer the blocks for computing coverage. For BED12 files, this uses the BlockCount, BlockStarts, and BlockEnds fields (i.e., columns 10,11,12). (-split) Strand Calculate coverage of intervals from a specific strand. With BED files, requires at least 6 columns (strand is column 6). (-strand) 5 prime Calculate coverage of 5' positions (instead of entire interval). (-5) 3 prime Calculate coverage of 3' positions (instead of entire interval). (-3) Max Combine all positions with a depth >= max into a single bin in the histogram. (-max) Scale Scale the coverage by a constant factor.Each coverage value is multiplied by this factor before being reported. Useful for normalizing coverage by, e.g., reads per million (RPM). Default is 1.0; i.e., unscaled. (-scale) Trackline Adds a UCSC/Genome-Browser track line definition in the first line of the output. (-trackline) Trackopts Writes additional track line definition parameters in the first line. (-trackopts) Block size Number of items to bundle in r-tree (-blockSize). Items per slot Number of data points bundled at lowest level (-itemsPerSlot). Uncompressed If set, do not use compression (-unc). Output Files Page: On this page you can select an output directory: