The workflow sample, described below, takes FASTQ files with paired-end Illumina reads and FASTQ file(s) with Oxford Nanopore reads and assembles these data de novo with SPAdes.
How to Use This Sample
If you haven't used the workflow samples in UGENEbefore, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "De novo Assemble Illumina PE and Nanopore Reads" can be found in the "NGS" section of the Workflow Designer samples.
Workflow Image
The opened workflow looks as follows:

Workflow Wizard
The wizard has 4 pages.
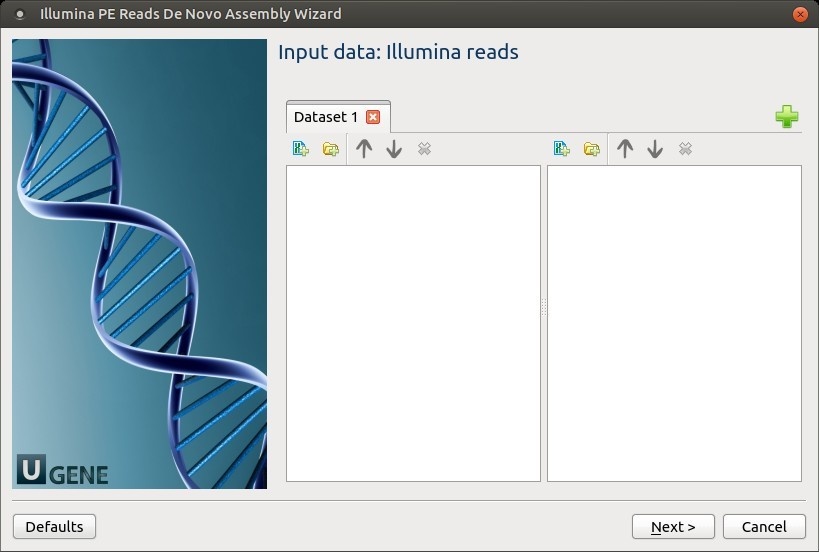
Input data: Illumina reads: On this page, files with Illumina reads must be set.
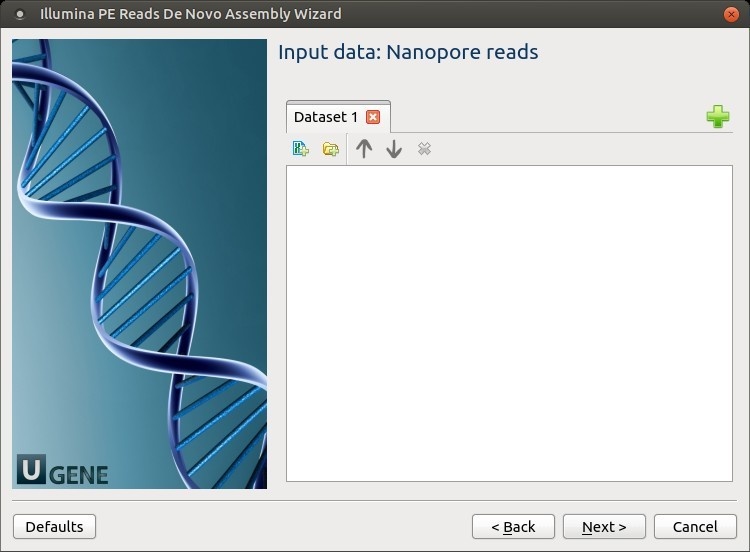
Input data: Nanopore reads: The Nanopore reads must be set on this page.
SPAdes settings: Default SPAdes parameters can be changed here.
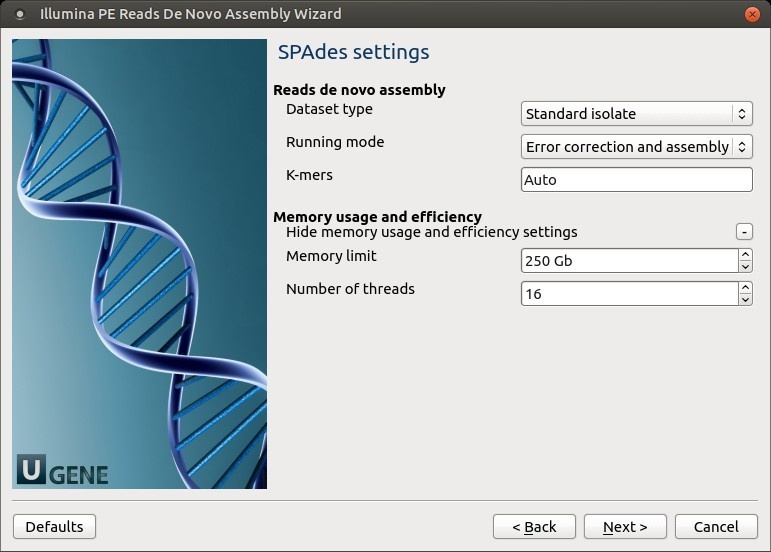
The following parameters are available:
Dataset type Select the input dataset type: standard isolate (the default value) or multiple displacement amplification (corresponds to --sc).
Running mode By default, SPAdes performs both read error correction and assembly. You can select leave one of only (corresponds to --only-assembler, --only-error-correction).
Error correction is performed using BayesHammer module in case of Illumina input reads andIonHammer in case of IonTorrent data. Note that you should not use error correction in case input reads do not have quality information(e.g. FASTA input files are provided).K-mers k-mer sizes (-k).
Output Files Page: On this page, you can select an output directory:
