To map Sanger sequencing reads to a reference use the Tools–> Sanger data analysis–> Map reads to reference main menu item. The following dialog appears:
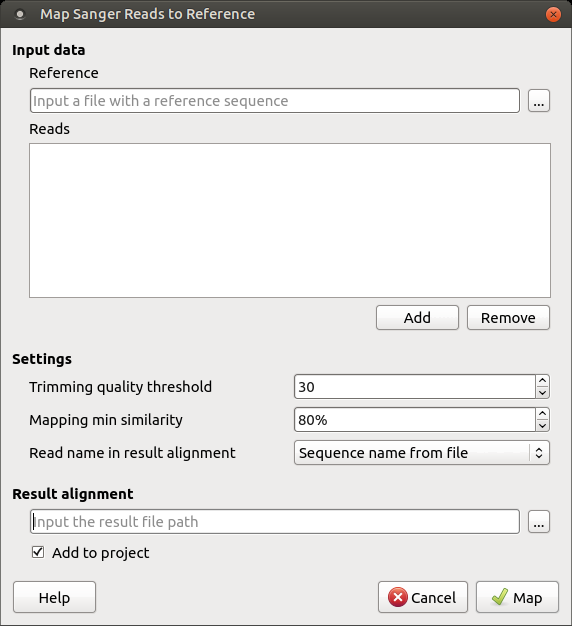
It is required to input:
Reference sequence — a file with a single DNA sequence of any supported file format (e.g. FASTA or GenBank). This parameter is mandatory.
Reads — a set of files in *.ab1 or *.scf format with the Sanger sequencing data to be mapped to the reference sequence. It is required to add at least one read. A read orientation (forward or reverse) will be automatically detected during the mapping.
During the mapping task execution, these data are processed as follows. First of all, to enhance the further mapping sensitivity, the low-quality ends of the reads are trimmed off. Then the reads are mapped using a UGENE original method that works in two steps: rough mapping of a read using BLAST+ and enhancement of the read alignment with the Smith-Waterman algorithm. After that the percentage similarity of the two sequences is calculated, that is the number of all edit operations (i.e., insertions, deletions, and substitutions) required to transform a read sequence into the corresponding region of the reference sequence. Reads that have low similarity with the reference sequence are filtered out.
One can configure the following parameters of the mapping task
Trimming quality threshold — all bases at the ends of the reads with quality lower than the specified value are trimmed. To skip the trimming, set this value to zero.
Mapping min similarity — all reads mapped to the reference with lower percentage similarity than the specified value are filtered out.
Read name in result alignment — reads in the resulting alignment can be named either by names of the sequences in the input files or by the input files names. Set this value to "File name", for example, if the sequences in the input *.ab1 files have the same name, this will help in distinguishing of the reads in the resulting alignment.
The resulting alignment is stored in a native UGENEDB format. One can set up the file location and name in the Result alignment field. Note that thereafter it is also possible to export data to standard alignment formats without chromatograms such as FASTA, ClustalW, etc.
To initiate the mapping task execution click the Map button in the dialog. Note that when the task is finished, the task statistics can be found in a report, available by clicking the corresponding notification:
