Use this workflow sample to process raw RNA-seq next-generation sequencing (NGS) data from the Illumina platform. The processing includes:
- Filtration:
- Filtering of the NGS short reads by the CASAVA 1.8 header;
- Trimming of the short reads by quality;
- [Optionally] Mapping:
- Mapping of the short reads to the specified reference sequence (the TopHat tool is used in the sample);
The result output of the workflow contains the filtered and merged FASTQ files. In case the TopHat mapping has been done, the result also contains the TopHat output files: the accepted hits BAM file and tracks of junctions, insertions and deletions in BED format. Other intermediate data files are also output by the workflow.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
What's Next?
Workflow Sample Location
The workflow sample "Raw DNA-Seq processing" can be found in the "NGS" section of the Workflow Designer samples.
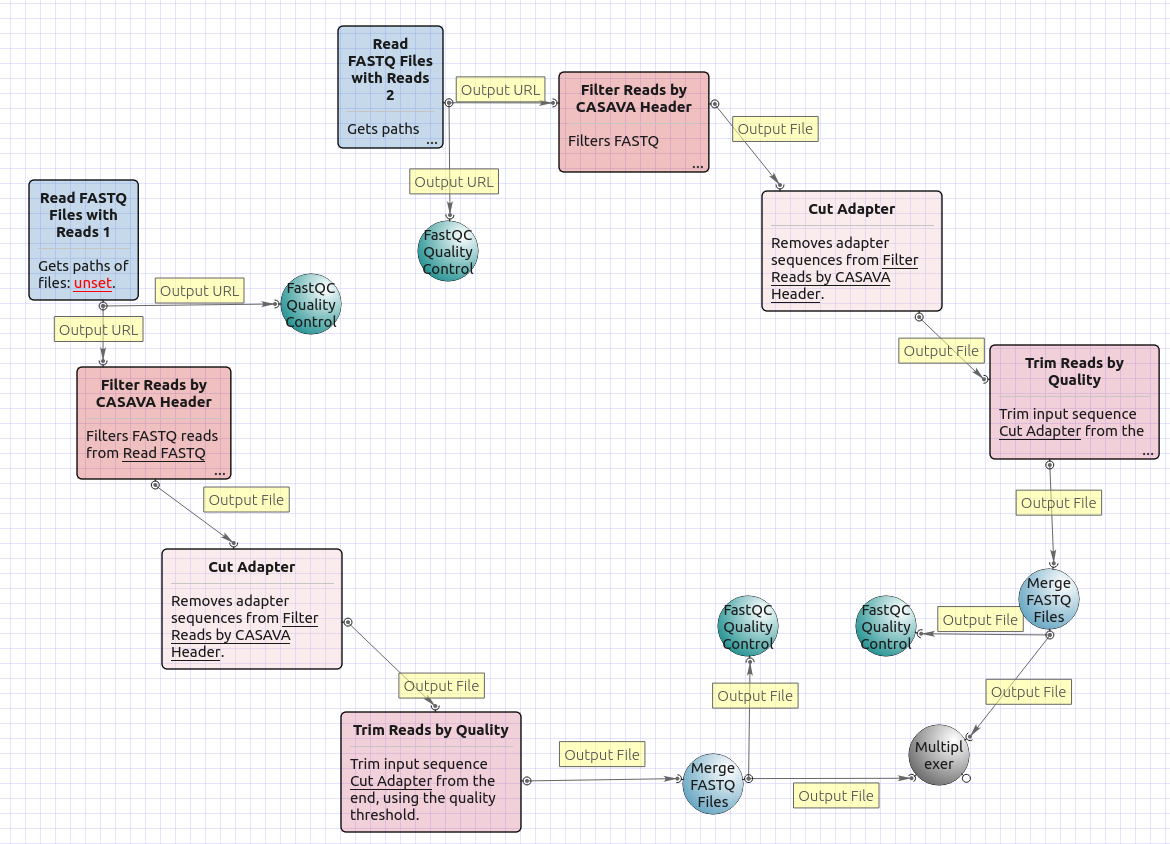
Workflow Image
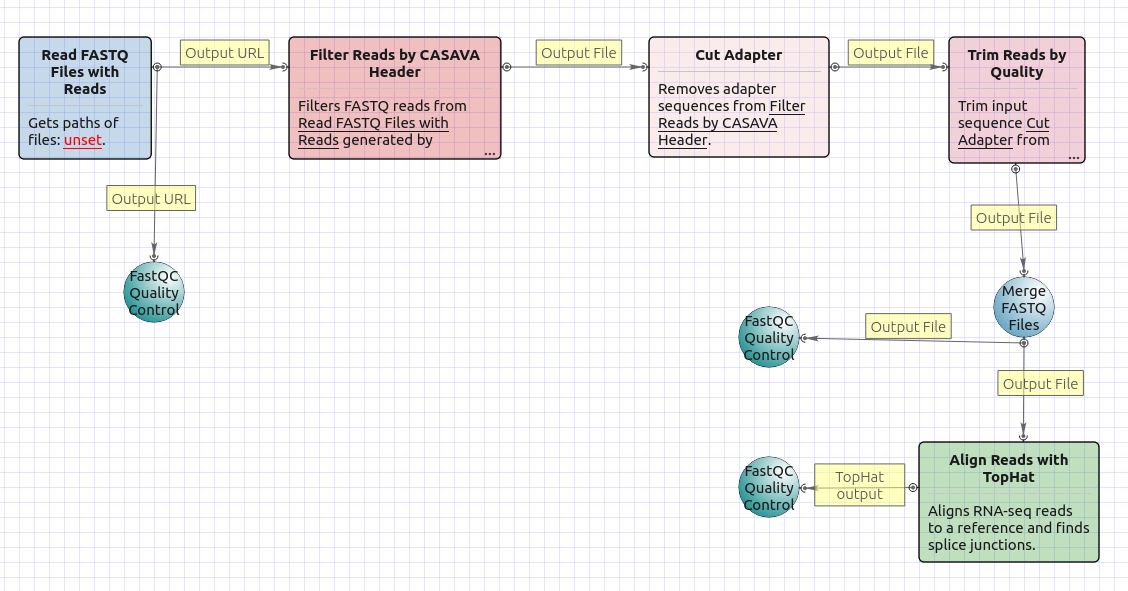
There are four versions of the workflow available. The workflow with mapping for single-end reads looks as follows:
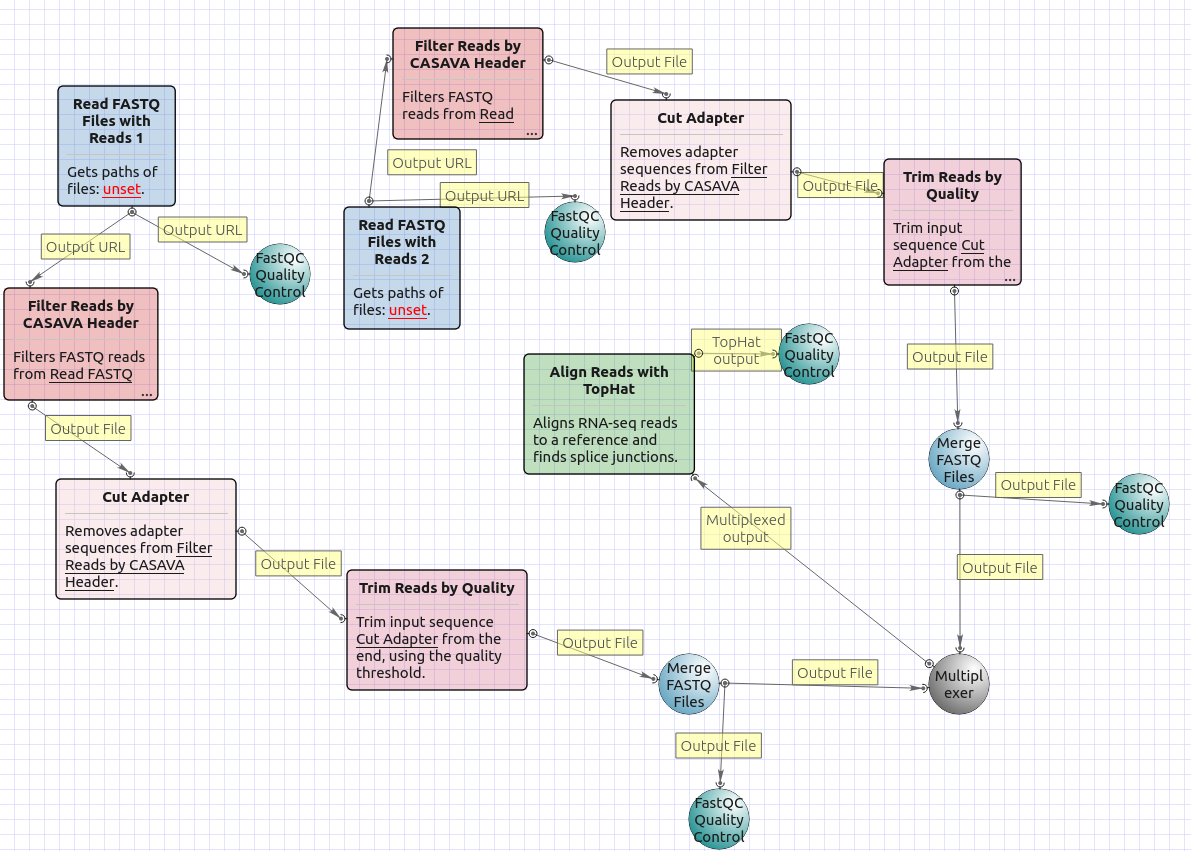
The workflow with mapping for paired-end short appearance is the following:
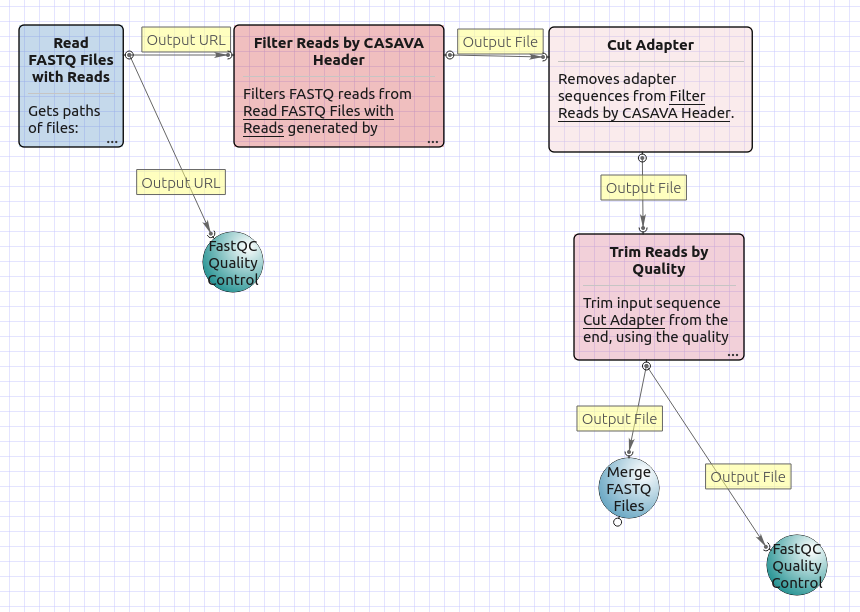
The workflow without mapping for single-end short appearance is the following:
The workflow without mapping for paired-end short appearance is the following:
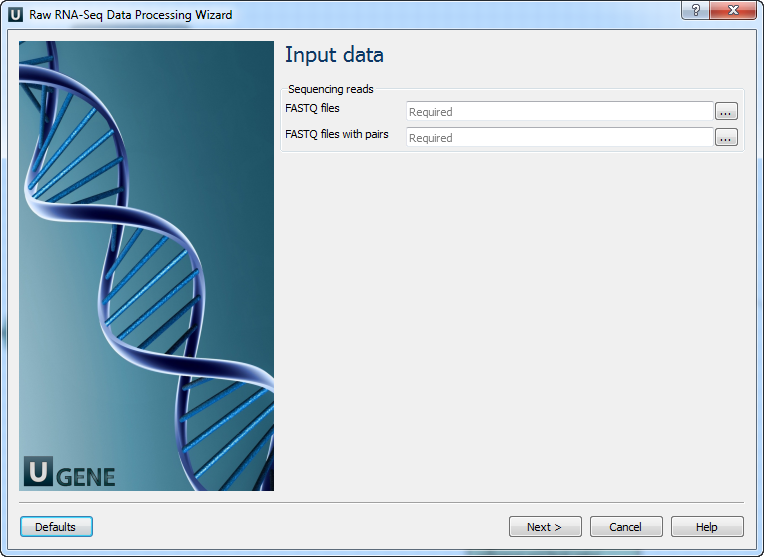
Workflow Wizard
The workflows have the similar wizards. The wizard for paired-end reads with mapping has 4 pages.
Input data: On this page you must input FASTQ file(s).
Pre-processing: On this page you can modify filtration parameters.
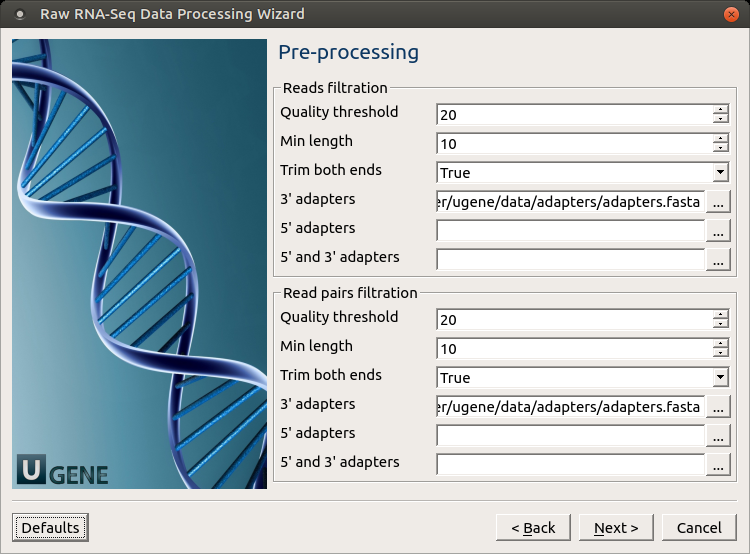
The following parameters are available for reads and reads pairs filtration:
Base quality Quality threshold for trimming. Reads length Too short reads are discarded by the filter. Trim both ends Trim the both ends of a read or not. Usually, you need to set True for Sanger sequencing and False for NGS 3' adapters A FASTA file with one or multiple sequences of adapter that were ligated to the 3' end. The adapter itself and anything that follows is trimmed. If the adapter sequence ends with the '$ character, the adapter is anchored to the end of the read and only found if it is a suffix of the read. 5' adapters A FASTA file with one or multiple sequences of adapters that were ligated to the 5' end. If the adapter sequence starts with the character ^, the adapter is 'anchored'.
An anchored adapter must appear in its entirety at the 5' end of the read (it is a prefix of the read). A non-anchored adapter may appear partially at the 5' end, or it may occur within the read.
If it is found within a read, the sequence preceding the adapter is also trimmed. In all cases, the adapter itself is trimmed.
5' and 3' adapters A FASTA file with one or multiple sequences of adapter that were ligated to the 5' end or 3' end. Mapping: On this page you must input reference and optionally modify advanced parameters.
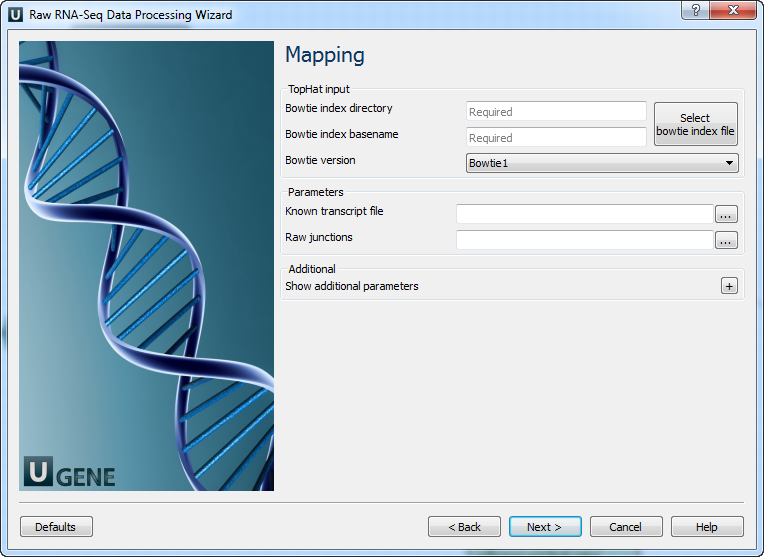
The following parameters are available:
Bowtie index directory The directory with the Bowtie index for the reference sequence. Bowtie index basename The basename of the Bowtie index for the reference sequence. Bowtie version Specifies which Bowtie version should be used. Known transcript file
A set of gene model annotations and/or known transcripts.
Raw junctions
The list of raw junctions.
Mate inner distance
Expected (mean) inner distance between mate pairs.
Mate standard deviation
Standard deviation for the distribution on inner distances between mate pairs.
Library type
Specifies RNA-seq protocol.
No novel junctions
Only look for reads across junctions indicated in the supplied GFF or junctions file. This parameter is ignored if Raw junctions or Known transcript file is not set.
Max multihints
Instructs TopHat to allow up to this many alignments to the reference for a given read, and suppresses all alignments for reads with more than this many alignments.
Segment length
Each read is cut up into segments, each at least this long. These segments are mapped independently.
Fusion search
Turn on fusion mapping.
Transcritome max hits
Only align the reads to the transcriptome and report only those mappings as genomic mappings.
Prefilter multihints
When mapping reads on the transcriptome, some repetitive or low complexity reads that would be discarded in the context of the genome may appear to align to the transcript sequences and thus may end up reported as mapped to those genes only. This option directs TopHat to first align the reads to the whole genome in order to determine and exclude such multi-mapped reads (according to the value of the Max multihits option).
Min anchor length
The anchor length. TopHat will report junctions spanned by reads with at least this many bases on each side of the junction. Note that individual spliced alignments may span a junction with fewer than this many bases on one side. However, every junction involved in spliced alignments is supported by at least one read with this many bases on each side.
Splice mismatches
The maximum number of mismatches that may appear in the anchor region of a spliced alignment.
Read mismatches
Final read alignments having more than these many mismatches are discarded.
Segment mismatches
Read segments are mapped independently, allowing up to this many mismatches in each segment alignment.
Solexa 1.3 quals
As of the Illumina GA pipeline version 1.3, quality scores are encoded in Phred-scaled base-64. Use this option for FASTQ files from pipeline 1.3 or later.
Bowtie version
specifies which Bowtie version should be used.
Bowtie -n mode
TopHat uses -v in Bowtie for initial read mapping (the default), but with this option, -n is used instead. Read segments are always mapped using -v option.
Bowtie tool path
The path to the Bowtie external tool.
SAMtools tool path
The path to the SAMtools tool. Note that the tool is available in the UGENE External Tool Package.
TopHat tool path
The path to the TopHat external tool in UGENE.
Temporary directory
The directory for temporary files.
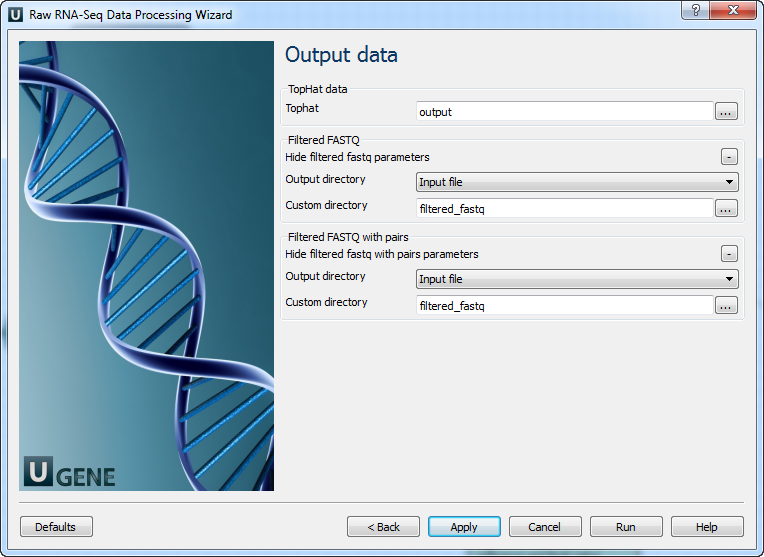
Output data: On this page you must input output parameters.