When you select the Tools ‣ Align to reference ‣ Align short reads item in the main menu, the Align Sequencing Reads dialog appears. Set value of the Align short reads method parameter to Bowtie 2. The dialog looks as follows:
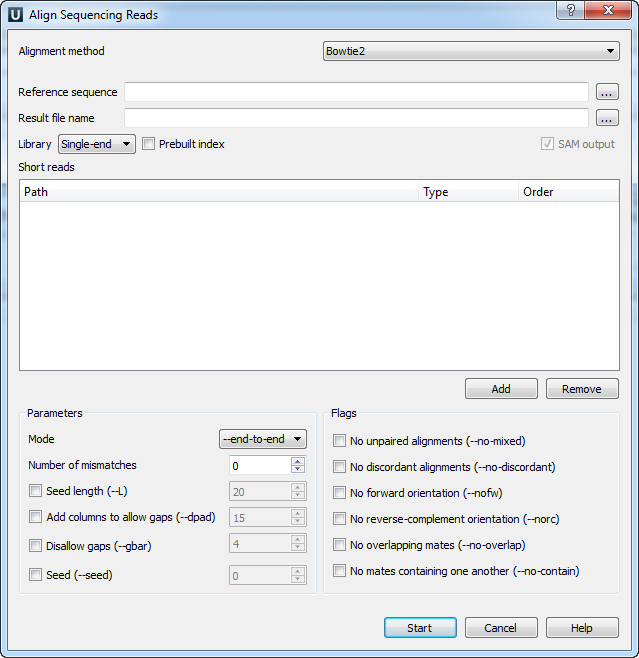
There are the following parameters:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in SAM format to write the result of the alignment into. This parameter is required.
Library - single-end or paired-end reads.
Prebuilt index — check this box to use an index file instead of a source reference sequence. The index is a set of 6 files with suffixes .1.ebwt, .2.ebwt, .3.ebwt, .4.ebwt, .rev.1.ebwt, and .rev.2.ebwt. The index is created during the alignment. Also you can build it manually.
SAM output — always save the output file in the SAM format (the option is disabled for Bowtie).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
You can also configure other parameters. They are the same as in the original Bowtie 2 (you can read detailed description of the parameters on the Bowtie 2 manual page).
Select one of the following alignment modes:
The --end-to-end alignment mode:
By default, Bowtie 2 performs end-to-end read alignment. That is, it searches for alignments involving all of the read characters. This is also called an "untrimmed" or "unclipped" alignment.
When the --local option is specified, Bowtie 2 performs local read alignment. In this mode, Bowtie 2 might "trim" or "clip" some read characters from one or both ends of the alignment if doing so maximizes the alignment score.
The following parameters are available:
Number of mismatches (--N) — sets the number of mismatches to allowed in a seed alignment during multiseed alignment. Can be set to 0 or 1. Setting this higher makes alignment slower (often much slower) but increases sensitivity.
Seed length (–L) — Sets the length of the seed substrings to align during multiseed alignment. Smaller values make alignment slower but more senstive.
Add columns to allow gaps (--dpad) — "Pads" dynamic programming problems by <int> columns on either side to allow gaps.
Disallow gaps (–gbar) — disallow gaps within <int> positions of the beginning or end of the read.
Seed (–seed) — use <int> as the seed for pseudo-random number generator.
The following flags are available:
No unpaired alignments (–no-mixed) — by default, when bowtie2 cannot find a concordant or discordant alignment for a pair, it then tries to find alignments for the individual mates. This option disables that behavior.
No discordant alignments (–no-discordant) — by default, bowtie2 looks for discordant alignments if it cannot find any concordant alignments. A discordant alignment is an alignment where both mates align uniquely, but that does not satisfy the paired-end constraints. This option disables that behavior.
No forward orientation (–nofw) — if --nofw is specified, bowtie2 will not attempt to align unpaired reads to the forward (Watson) reference strand.
No reverse-complement orientation (–norc) — if --norc is specified, bowtie2 will not attempt to align unpaired reads against the reverse-complement (Crick) reference strand.
No overlapping mates (–no-overlap) — if one mate alignment overlaps the other at all, consider that to be non-concordant.
No mates containing one another (–no-contain) — if one mate alignment contains the other, consider that to be non-concordant.
Select the required parameters and press the Start button.