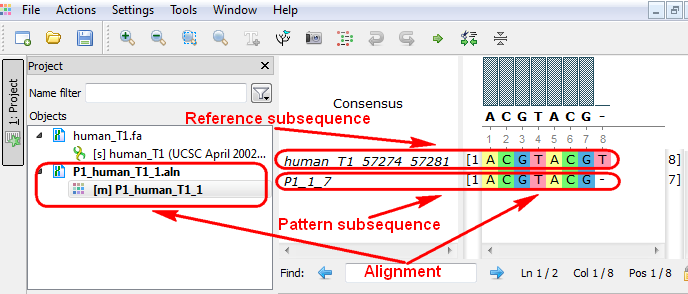
The Smith-Waterman Search plugin adds a complete implementation of the Smith-Waterman algorithm to UGENE.
To use the plugin open a nucleotide or protein sequence in the Sequence View and select the Analyze ‣ Find pattern [Smith-Waterman] item in the context menu. The Smith-Waterman Search dialog appears:
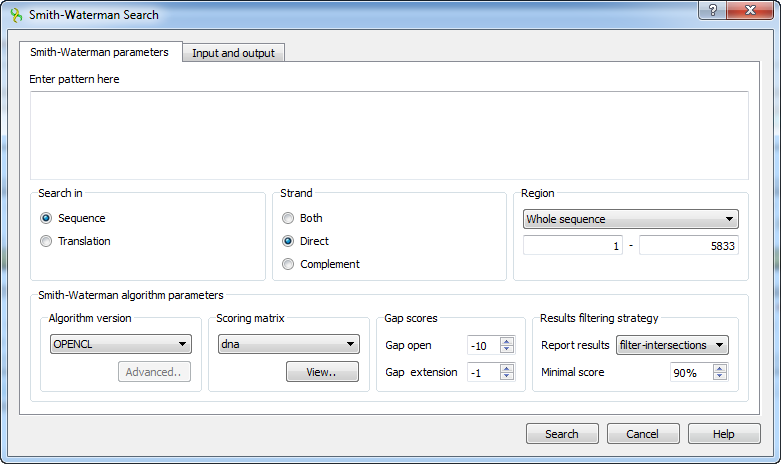
First of all you need to specify the pattern to search for. The rest parameters are optional:
Search in — select either to search in the sequence or in its translation.
Strand — select the strand to search in: direct, complementary or both strands.
Region — specifies the region of the sequence that will be used to search for the pattern. By default, if a subsequence has been selected when the dialog has been opened, then the selected subsequence is searched for the pattern. Otherwise, the whole sequence is used. You can also input a custom range.
Algorithm version — version of the algorithm implementation. Non-classic versions produce the same results as classic but much faster. To use these optimizations our system must support these capabilities.
- Classic 2
- SSE2
- CUDA
- OPENCL
Scoring matrix — can be chosen from a bunch of matrices supplied with UGENE. To view a matrix selected click the View button.
Gap open — penalty for opening a gap.
Gap extension — penalty for extending a gap
Report results — simple heuristic which allows to filter intersected hits. If it is set to none, the algorithm may report large set of almost identical results in the same region.
Minimal score — another simple heuristic which measures sequences similarity. It is more convenient than using some abstract scores. If set to 100%, the algorithm will search for exact substring match.
The results of the search are saved as annotations or as multiple alignment. To set the saving parameters go to the Input and output tab of the dialog.
If you want to save the results as annotations input the annotations saving parameters (Annotation name, Group name, a file to save the annotation to). Also you can add qualifier with corresponding pattern subsequences to result annotations. Check the corresponding checkbox for it.
If you want to save the results as multiple alignment select the following parameters:
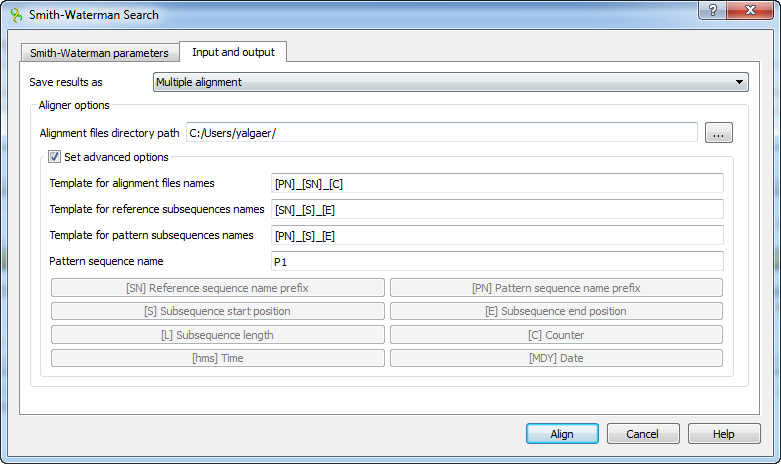
Here you can select a file to save the alignment to (Alignment files directory path parameter).
Using the Set advanced options checkbox you can select the saving options.
You can set the different templates for files names: create your own or create by using the following: [E] — adds a subsequence end position, [hms] — adds a time, [MDY] — adds a date, [S] — adds a subsequence start position, [L] — adds a subsequence length, [SN] — adds a reference sequence name prefix, [PN] — adds a pattern sequence name prefix, [C] — adds a counter.
You can create templates for alignment files names, reference subsequence names, pattern subsequence names and for pattern sequence name: