To make a request to a local BLAST database do the following:
- If you’re using BLAST open Tools ‣ BLAST ‣ BLAST Search.
- If you’re using BLAST+ open Open Tools ‣ BLAST ‣ BLAST+ Search.
If there is a sequence opened you can also initiate the request to a local BLAST database from the Sequence View:
- If you’re using BLAST select the Analyze ‣ Query with BLAST item in the context menu or in the Actions main menu.
- If you’re using BLAST+ select the Analyze ‣ Query with BLAST+ item in the context menu or in the Actions main menu.
The Request to local BLAST database dialog will appear:
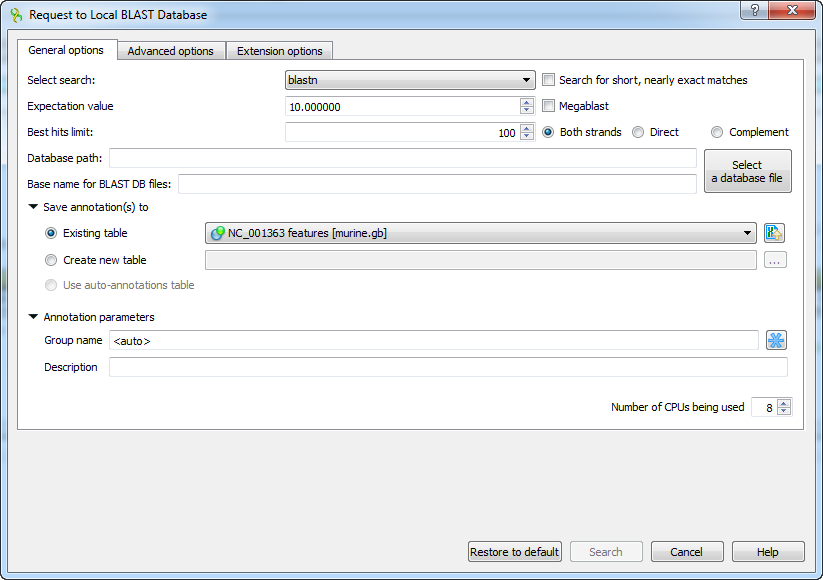
The following general options are available:
Select search - here you should select the tool you would like to use. If the query sequence is a nucleotide sequence then blastn, blastx and tblastx items are available. For a protein sequence the items are blastp and tblastn.
Expectation value - this option specifies the statistical significance threshold for reporting matches against database sequences. Lower expect thresholds are more stringent, leading to fewer chance matches being reported.
Culling limit - the maximum number of hits that will be shown (not equal to number of annotations). The maximum availablle number is 5000.
Search for short, nearly exact matches - automatically adjusts the word size and other parameters to improve results for short queries.
Megablast - select this option to compare query with closely related sequences. It works best if the target percent identity is 95% or more, but it is very fast.
Database path - path to the database files.
Base name for BLAST DB files - base name for the BLAST database files.
You can see the description of the annotation saving parameters here.
The following advanced parameters are available:
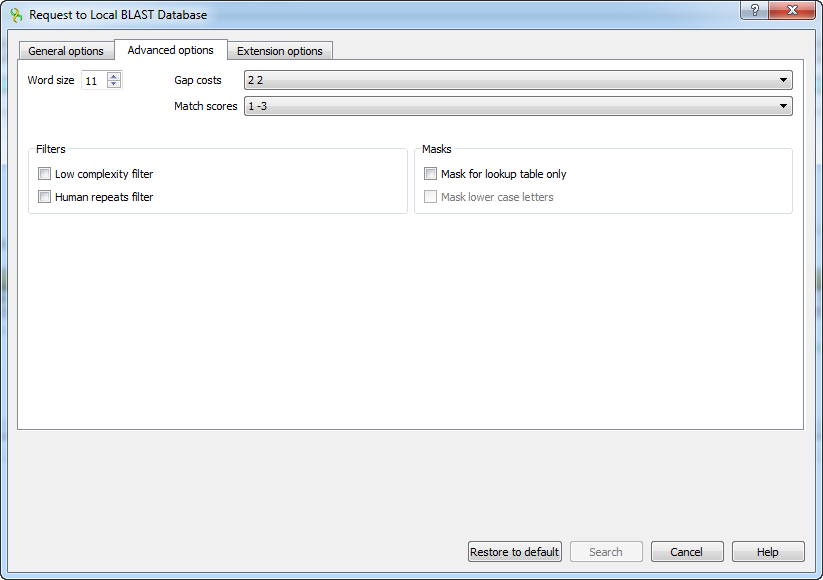
Word size - the size of the subsequence parameter for the initiated search.
Gap costs - costs to create and extend a gap in an alignment. Increasing the Gap costs will result in alignments which decrease the number of Gaps introduced.
Match scores - reward and penalty for matching and mismatching bases.
Filters - filters for regions of low compositional complexity and repeat elements of the human’s genome.
Masks for lookup table only — this option masks only for purposes of constructing the lookup table used by BLAST so that no hits are found based upon low-complexity sequence or repeats (if repeat filter is checked).
Mask lower case letters — with this option selected you can cut and paste a FASTA sequence in upper case characters and denote areas you would like filtered with lower case.
The view of the Advanced options tab depends on the selected search. For the blastn search it looks like on the picture above. When the blastx search is selected in the general options, the view of the Advanced options tab is the following:
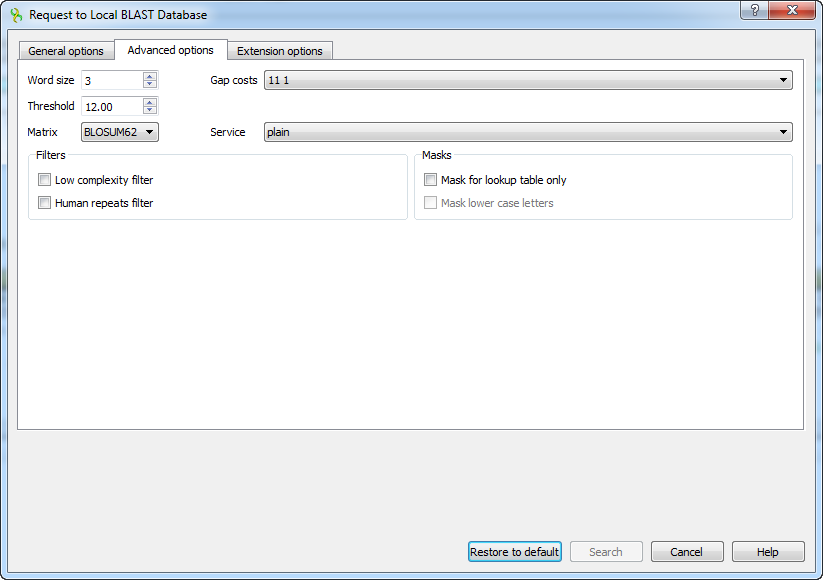
As you can see there is no Match scores option, but there are Threshold, Matrix, Composition-based statistics and Service options.
Threshold - threshold for extending hits.
Matrix — key element in evaluating the quality of a pair-wise sequence alignment is the “substitution matrix”, which assigns a score for aligning any possible pair of residues.
Service — blastp service which needs to be performed: plain, psi or phi.
Composition-based statistics - composition-based statistics.
When the tblastx search is selected in the general options, the view of the Advanced options tab is the following:
The following extension options are available:
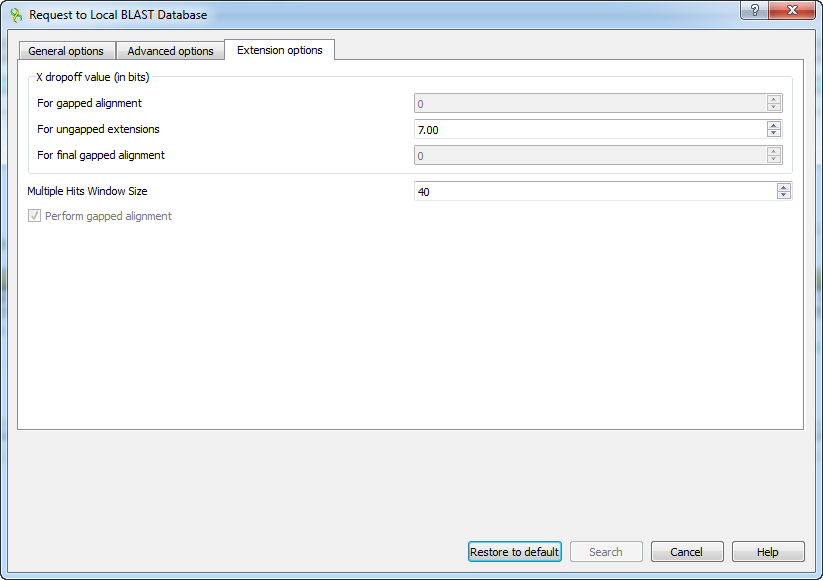
For gapped alignment - X dropoff value (in bits) for gapped alignment.
For ungapped alignment - X dropoff value (in bits) for ungapped alignment.
For final gapped alignment - X dropoff value (in bits) for final gapped alignment.
Multiple hits window size - multiple hits window size.
Perform gapped alignment - performs gapped alignment.