The DAS annotator finds similar protein sequence using remote BLAST. Using IDs of sequences found loads annotation for DAS sources. Nucleotide sequences are skipped.To annotate with DAS use the DAS Annotations tab of the Options Panel:
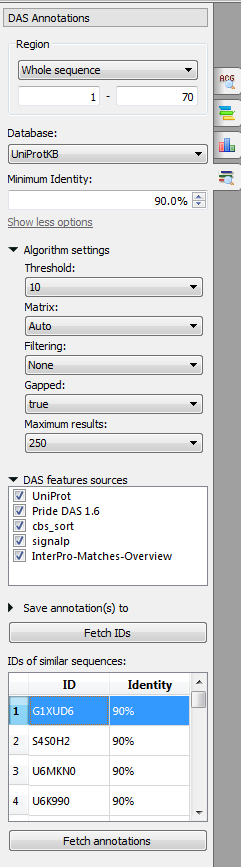
The following parameters are available:
Region - region for finding.
Database - database against which the search is performed: UniProtKB or clusters of sequences with 100%, 90% or 50% identity.
Minimum identity - minimum identity of a BLAST result and an input sequence.
Algorithm settings:
Threshold - the expectation value (E) threshold is a statistical measure of the number of expected matches in a random database. The lower the e-value, the more likely the match is to be significant.
Matrix - the matrix assigns a probability score for each position in an alignment.
Filtering - low-complexity regions (e.g. stretches of cysteine in Q03751, or hydrophobic regions in membrane proteins) tend to producespurious, insignificant matches with sequences in the database which have the same kind of low-complexity regions, but are unrelated biologically. If 'Filter low complexity regions' is selected, the query sequence will be run through the program SEG, and all amino acids in low-complexity regions will be replaced by X's.
Gapped - this will allow gaps to be introduced in the sequences when the comparison is done.
Maximum results - limits the number of returned alignments.
DAS features sources - the DAS sources to read features from.
Save annotations to - allows to select the annotation table.
IDs of similar sequences - the list of the IDs of the similar sequences.
Select the parameters and click on the Fetch IDs button. The sequences will appear in the IDs of similar sequences table. To fetch annotations select it (to select several IDs use the Ctrl or Shift buttons) and click on the Fetch annotations button. The annotations will appear.