When you select the Tools ‣ Align to reference ‣ Align short reads item in the main menu, the Align Sequencing Reads dialog appears. Set value of the Align short reads method parameter to BWA-MEM. The dialog looks as follows:
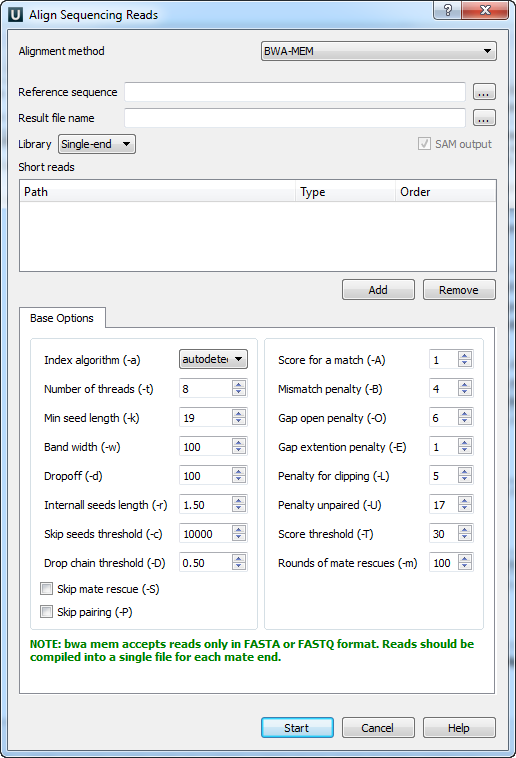
There are the following parameters:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in SAM format to write the result of the alignment into. This parameter is required.
Prebuilt index — check this box to use an index file instead of a source reference sequence. Also you can build it manually.
SAM output — always save the output file in the SAM format (the option is disabled for BWA).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
You can also configure other parameters.
Index algorithm (-a) — algorithm for constructing BWA index.
It implements three different algorithms:
- is — designed for short reads up to ~200bp with low error rate (<3%). It does gapped global alignment w.r.t. reads, supports paired-end reads, and is one of the fastest short read alignment algorithms to date while also visiting suboptimal hits.
- bwtsw — is designed for long reads with more errors. It performs heuristic Smith-Waterman-like alignment to find high-scoring local hits. Algorithm implemented in BWA-SW. On low-error short queries, BWA-SW. is slower and less accurate than the is algorithm, but on long reads, it is better.
- div — does not work for long genomes.
Number of threads (-t) — number of threads.
Min seed length (-k) — minimum seed length. Matches shorter than INT will be missed. The alignment speed is usually insensitive to this value unless it significantly deviates 20.
Band width (-w) — band width. Essentially, gaps longer than INT will not be found. Note that the maximum gap length is also affected by the scoring matrix and the hit length, not solely determined by this option.
Dropoff (-d) — off-diagonal X-dropoff (Z-dropoff). Stop extension when the difference between the best and the current extension score is above |i-j|*A+INT, where i and j are the current positions of the query and reference, respectively, and A is the matching score. Z-dropoff is similar to BLAST’s X-dropoff except that it doesn’t penalize gaps in one of the sequences in the alignment. Z-dropoff not only avoids unnecessary extension, but also reduces poor alignments inside a long good alignment.
Internall seeds length (-r) - trigger re-seeding for a MEM longer than minSeedLen*FLOAT. This is a key heuristic parameter for tuning the performance. Larger value yields fewer seeds, which leads to faster alignment speed but lower accuracy.
Skip seeds threshold (-c) - discard a MEM if it has more than INT occurence in the genome. This is an insensitive parameter.
Drop chain threshold (-D) - drop chains shorter than FLOAT fraction of the longest overlapping chain.
Rounds of mate rescues (-m) - perform at most INT rounds of mate rescues for each read.
Skip mate rescue (-S) - skip mate rescue.
Skip pairing (-P) - in the paired-end mode, perform SW to rescue missing hits only but do not try to find hits that fit a proper pair.
Score for a match (-A) - matching score.
Mismatch penalty (-B) - mismatch penalty. The sequence error rate is approximately: {.75 * exp[-log(4) * B/A]}.
Gap open penalty (-O) - gap open penalty.
Gap extention penalty (-E) - gap extension penalty. A gap of length k costs O + k*E (i.e. Gap open penalty is for opening a zero-length gap).
Penalty for clipping (-L) - clipping penalty. When performing SW extension, BWA-MEM keeps track of the best score reaching the end of query. If this score is larger than the best SW score minus the clipping penalty, clipping will not be applied. Note that in this case, the SAM AS tag reports the best SW score; clipping penalty is not deducted.
Penalty unpaired (-U) - penalty for an unpaired read pair. BWA-MEM scores an unpaired read pair as scoreRead1+scoreRead2-INT and scores a paired as scoreRead1+scoreRead2-insertPenalty. It compares these two scores to determine whether we should force pairing.
Score threshold (-T) - don’t output alignment with score lower than score threshold. This option only affects output.
Select the required parameters and press the Start button.