To align two sequences go to the Pairwise Alignment tab of the Options Panel:
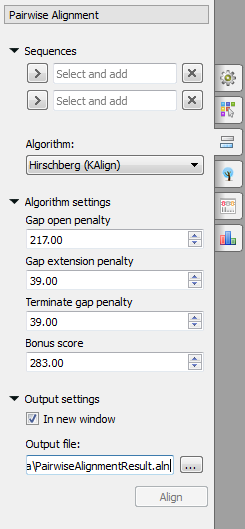
Select two sequence from the original alignment, select the parameters and click on the Align button. The following parameters are available:
Algorithm - algorithm of the pairwise alignment. There are two algorithms:
Hirschberg (KAlign) - algorithm has the following parameters:
Gap open penalty - indicates the penalty applied for opening a gap. The penalty must be negative.
Gap extension penalty - indicates the penalty applied for extending a gap.
Terminate gap penalty - the penalty to extend gaps from the N/C terminal of protein or 5'/3' terminal of nucleotide sequences.
Bonus score - a bonus score that is added to each pair of aligned residues.
Smith-Waterman - the following parameters are available:
Algorithm version - version of the algorithm implementation. Non-classic versions produce the same results as classic but much faster. To use these optimizations our system must support these capabilities: OPENCL, SSE2 or SW_classic.
Scoring matrix - scoring matrix.
Gap open penalty - penalty for opening a gap.
Gap extension penalty — penalty for extending a gap.
Output settings - settings of the otput file.