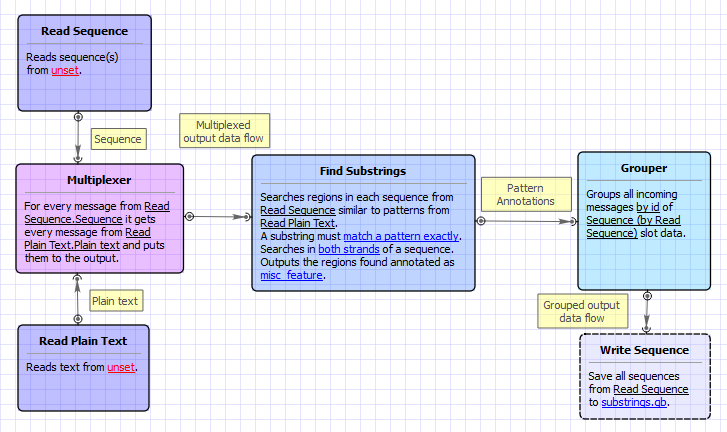
This sample workflow shows how to find substrings in input sequences, annotate them, and merge the found substring annotations with the original sequence annotations.
The steps of the workflow are these:
- The workflow reads sequences from the input sequence files (e.g. GenBank). The input data may also contain the annotations, associated with the sequences.
- The workflow reads text strings (patterns) from the input text files.
- The data are multiplexed using the Multiplexer element. Multiplexing rule "1 to many" is used, so each input sequence is concatenated with each pattern. The concatenating results are sent to the Find Substrings element.
- The Find Substrings element searches for the specified patterns in each sequence.
- The next element Grouper merges annotations, read for the sequence in the Read Sequence element, with annotations, found for the sequence by the Find Substrings element. A sequence ID is used to group the appropriate sets of annotations.
- And finally, the data are written to the output file ("substrings.gb" , by default).
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Find Substrings at Sequences" can be found in the "Data Merging" section of the Workflow Designer samples.
Workflow Image
The workflow looks as follows:
Workflow Wizard
The wizard has 3 pages.
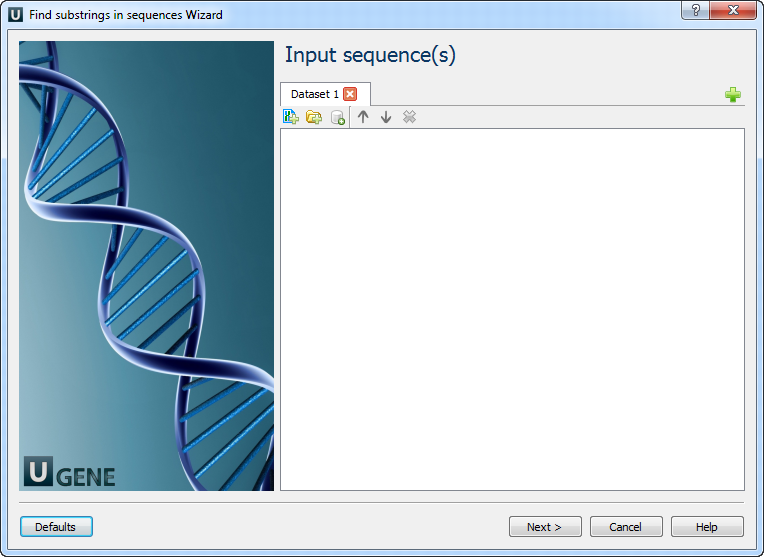
Input sequence(s): On this page you must input sequence(s).
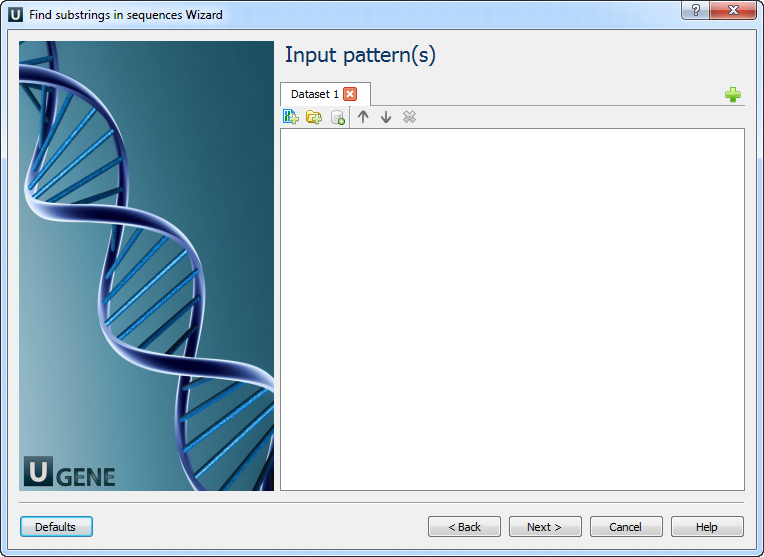
Input pattern(s): On this page you must input pattern(s).
Find substrings: On this page you can modify search and output parameters.
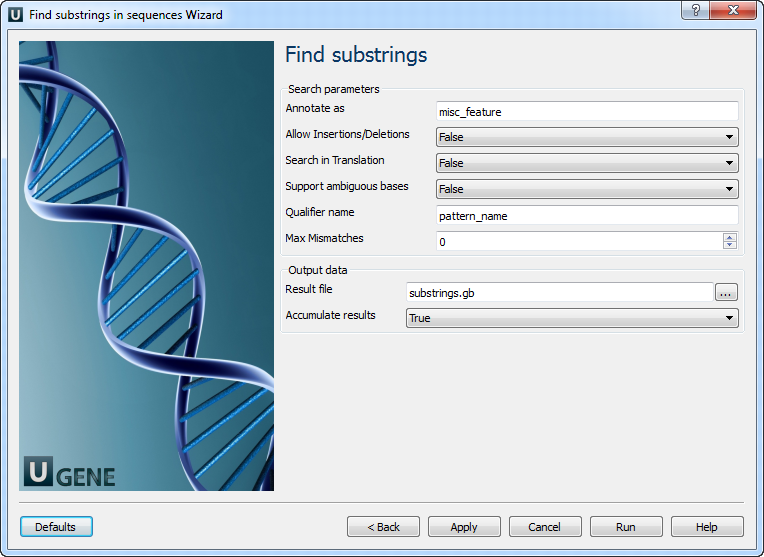
The following parameters are available:
Annotate as Name of the result annotations. Allow Insertions/Deletions Takes into account possibility of insertions/deletions when searching. By default substitutions are only considered. Search in Translation Translates a supplied nucleotide sequence to protein and searches in the translated sequence. Support ambiguous bases
Performs correct handling of ambiguous bases. When this option is activated insertions and deletions are not considered.
Qualifier name
Name of qualifier in result annotations which is containing a pattern name.
Max Mismatches
Maximum number of mismatches between a substring and a pattern.
Result file
Location of output data file. If this attribute is set, slot "Location" in port will not be used.
Accumulate results
Accumulate all incoming data in one file or create separate files for each input.In the latter case, an incremental numerical suffix is added to the file name.