This workflow builds a new profile HMM from input alignment, calibrates the HMM and saves to a file. Then runs a test HMM search over sample sequence and saves test results to Genbank file.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Build HMM from Alignment and test it" can be found in the "HMMER" section of the Workflow Designer samples.
Workflow Image
The workflow looks as follows:
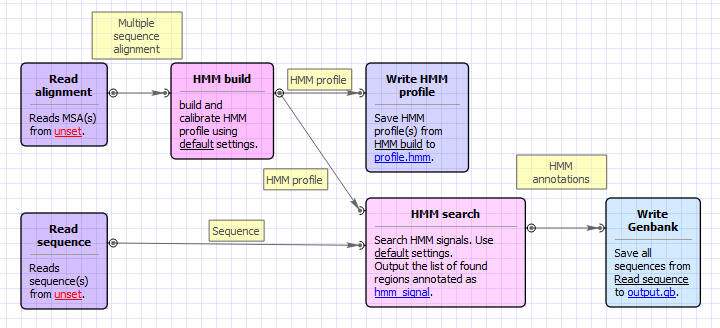
Workflow Wizard
The wizard has 4 pages.
Input MSA(s): On this page you must input MSA(s).
Input sequence(s): On this page you must input sequence(s).
HMM build: On this page you can modify HMM build parameters.
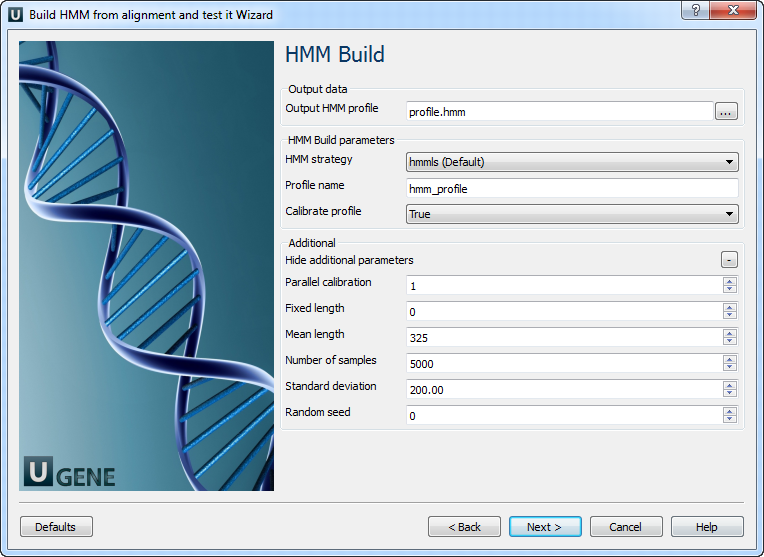
The following parameters are available:
Output HMM profile Location of output data file. If this attribute is set, slot "Location" in port will not be used. HMM strategy Specifies kind of alignments you want to allow. Profile name Descriptive name of the HMM profile. Calibrate profile
Enables/disables optional profile calibration.
An empirical HMM calibration costs time but it only has to be done once per model, and can greatly increase the sensitivity of a database search.
Parallel calibration
Number of parallel threads that the calibration will run in.
Fixed length
Fix the length of the random sequences to , where is a positive (and reasonably sized) integer. The default is instead to generate sequences with a variety of different lengths, controlled by a Gaussian (normal) distribution.
Mean length
Mean length of the synthetic sequences, positive real number. The default value is 325.
Number of samples
Number of synthetic sequences. If is less than about 1000, the fit to the EVD may fail. Higher numbers of will give better determined EVD parameters. The default is 5000; it was empirically chosen as a tradeoff between accuracy and computation time.
Standard deviation Standard deviation of the synthetic sequence length. A positive number. The default is 200. Note that the Gaussian is left-truncated so that no sequences have lengths
Random seed The random seed, where is a positive integer. The default is to use time() to generate a different seed for each run, which means that two different runs of hmmcalibrate on the same HMM will give slightly different results. You can use this option to generate reproducible results for different hmmcalibrate runs on the same HMM. HMM search: On this page you can modify HMM search and output parameters.
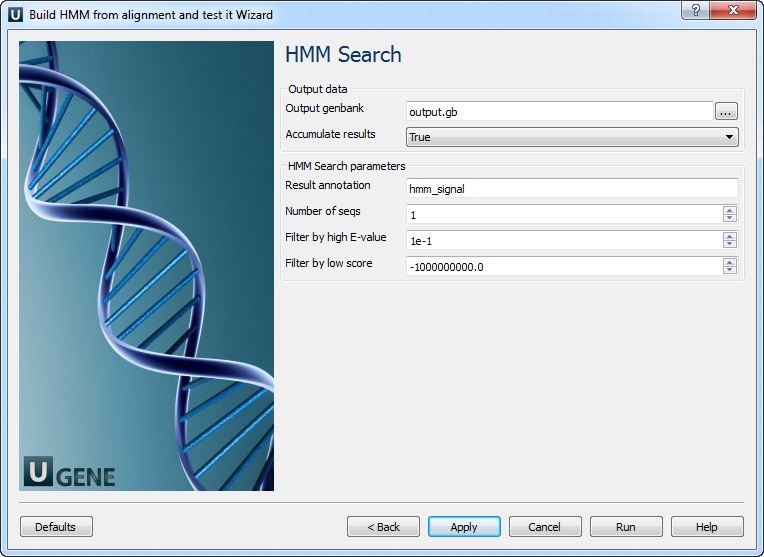
The following parameters are available:
Output genbank Location of output data file. If this attribute is set, slot "Location" in port will not be used. Accumulate results Accumulate all incoming data in one file or create separate files for each input.In the latter case, an incremental numerical suffix is added to the file name. Result annotation A name of the result annotations. Number of seqs
Calculate the E-value scores as if we had seen a sequence database of sequences. Filter by high E-value
E-value filtering can be used to exclude low-probability hits from result. Filter by low score
Score based filtering is an alternative to E-value filtering to exclude low-probability hits from result.