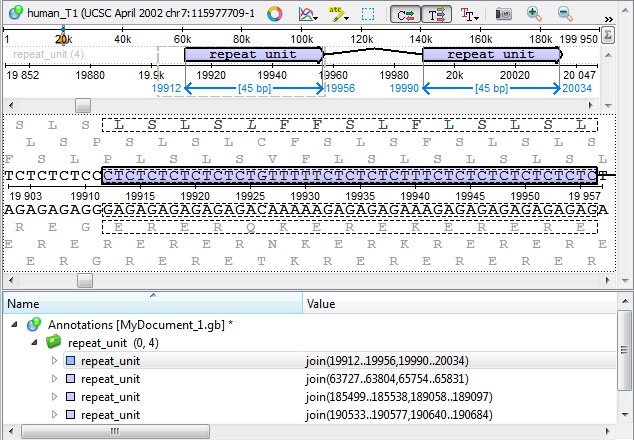
The Repeats Finding algorithm allows users to find repeated subsequences in accordance with the specified parameters.
Open a DNA sequence in the Sequence View. There are two ways to open the Find Repeats dialog:
- By clicking on the toolbar button:

- By selecting the Analyze ‣ Find repeats... context menu item:
The dialog following dialog appears:
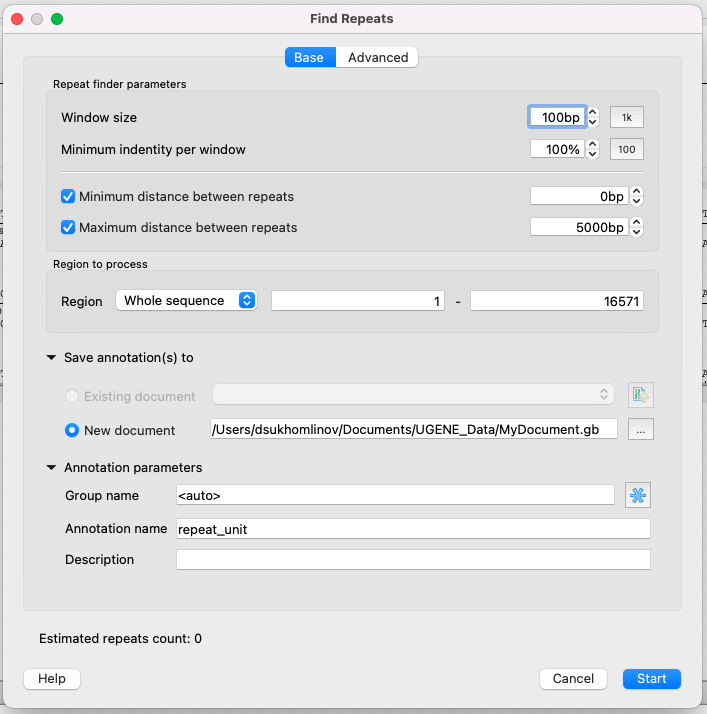 Parameters of the Base page
Parameters of the Base page
Repeat finder parameters
- Window size - to put it simply, this is the minimum length of a single repeat.
- Minimum identity per window - the minimum identity of repeated subsequences (in persentage).
- Minimum/Maximum distance between repeats - it is obvious.
Region to process
- Region - specifies the region of the sequence that will be used to search for repeats. By default, if a subsequence has been selected when the dialog has been opened, then the selected subsequence is searched for the pattern. Otherwise, the whole sequence is used. You can also input a custom range.
Save annotations
- The result annotations will be saved using the annotations saving parameters (Annotation name, Group name, Annotation type, Description and a file to save the annotation to).
Parameters of the Advansed page
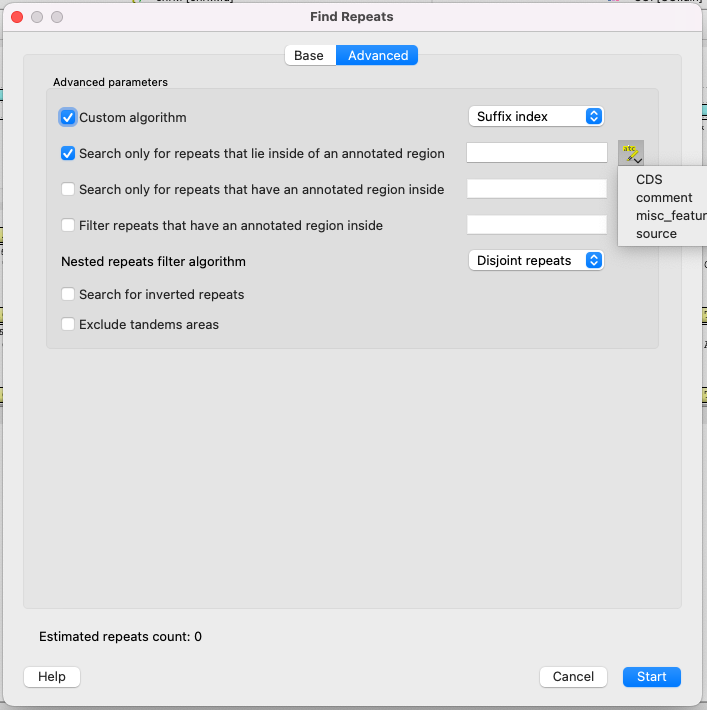
Advansed parameters:
- Custom algorithm - choose calculation algorithm. It will not affect results, but may affect the calculation time:
- Auto (recommended) - the most suitable algorithm will be choosen automatically.
- Suffix index - the search window moves along the entire sequence, each time adding one character at the end and removing one character at the beginning. The algorithm calculation time directly depends on the length of the sequence.
- Diagonals - the Divide-and-conquer algorithm, the subsequences are split in half recursively. More applicable for large sequences.
- Search only for repeats that lie inside of an annotated region.
- Search only for repeats that have an annotated region inside.
- Filter (get rid of) repeats that have an annotated region inside.
- Nested repeats filter algorithm - how to process nested repeats:
- No filtering - all possible repeats will be found.
- Disjoint repeats - repeats in which the left and right parts intersect will be filtered out.
- Unique repeats - only pairs of repeats that occur only once in the sequence (even if repeats intersect each other).
- Search for inverted repeats - search for repeats, which are located on differend DNA strands.
- Exclude tandem areas - exclude repeats if there are three or more of them and they come in a row (use the Tandem Repeats Finding feature for this case).
Results
The found repeats are saved and displayed as annotations to the DNA sequence: