CAP3 (CONTIG ASSEMBLY PROGRAM Version 3) is a sequence assembly program for small-scale assembly with or without quality values. Click this link to open CAP3 homepage. CAP3 is embedded as an external tool into UGENE.
Open Tools ‣ Sanger data analysis submenu of the main menu.

Select the Reads de novo assembly (with CAP3) item to use the CAP3.
The Contig Assembly With CAP3 dialog appears.
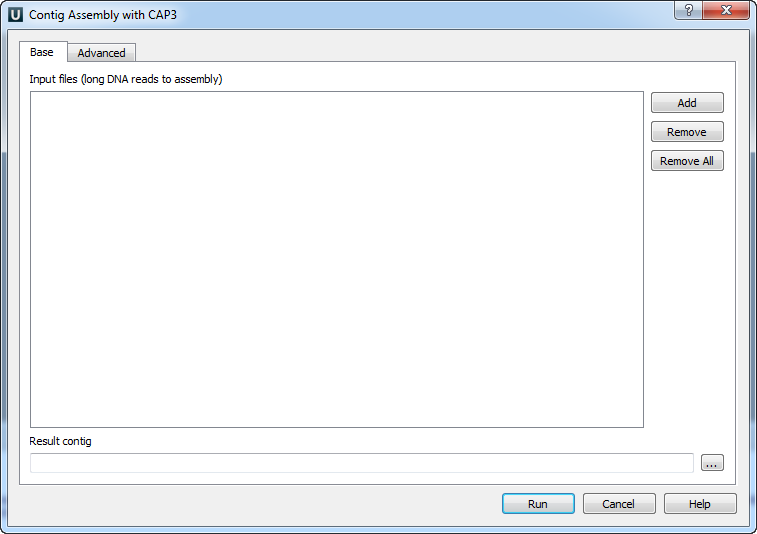
You can add or remove input files using Add and Remove buttons. To remove all files click the Remove all button. Input files are files with a long DNA reads in FASTA, FASTQ, SCF or ABI formats. At least one input file should be added. Input a Result contig name and press the Run button. CAP3 produces assembly results in the ACE file format (”.ace”). The file contains one or several contigs assembled from the input reads.
The quality scores for FASTA sequences can be provided in an additional file. The file must be located in the same folder as the original sequences and have the same name as FASTA file, but another extension: .qual.
Also you can change the following advanced parameters:
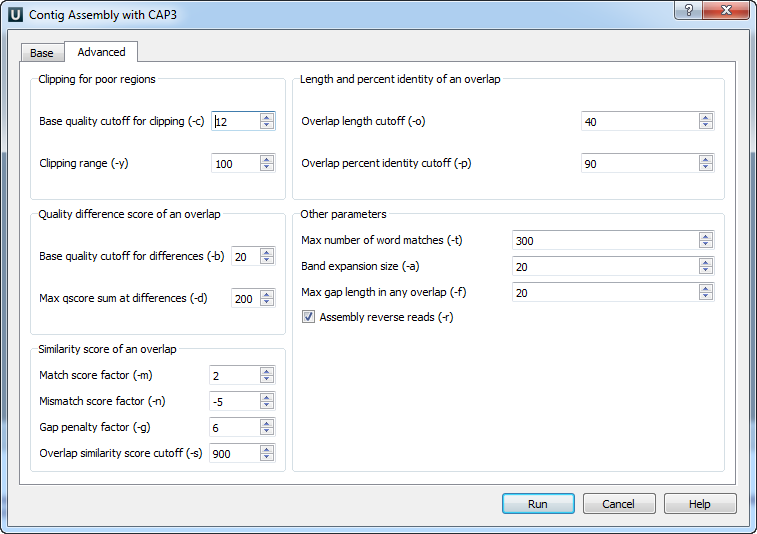
Clipping for poor regions parameters:
Clipping of a poor end region of a read is controlled by parameters Base quality cutoff for clipping (-c) (the specified value should be more than 5), and Clipping range (-y) (the specified value should be more than 5).
Quality difference score of an overlap parameters:
Base quality cutoff for differences (-b) — if an overlap contains a difference at bases of quality values q1 and q2, then the score at the difference is max(0, min(q1, q2) - b), where b is the specified value. The specified value should be more than 15. The difference score of an overlap is the sum of scores at each difference.
Max qscore sum at differences (-d) — remove an overlap if its difference score is greater than the specified value. The specified value should be more than 20.
Similarity score of an overlap parameters:
The following parameters are used to calculate the similarity score of an overlapping alignment:
Match score factor (-m) — a match at bases of quality values q1 and q2 is given a score of m * min(q1, q2), where m is the specified value. The specified value should be more than 0.
Mismatch score factor (-n) — a mismatch at bases of quality values q1 and q2 is given a score of n * min(q1, q2), where n is the specified value. The specified value should be less than 0.
Gap penalty factor (-g) — a base of quality value q1 in a gap is given a score -g * min(q1, q2), where g is the specified value; q2 is the quality value of the base in the other sequence right before the gap. The specified value should be more than 0.
The similarity score is caclulated as the sum of scores of each match, each mismatch and each gap. Based on this value and the following value some overlaps are removed:
Overlap similarity score cutoff (-s) — remove overlaps with similarity scores less than the specified value. The specified value should be more than 250.
Length and percent identity of an overlap parameters:
Overlap length cutoff (-o) — minimum length of an overlap (in base pairs). The specified value should be more than 15 base pairs.
Overlap percent identity cutoff (-p) — minimum percent identity of an overlap. The specified value should be more than 65%.
Other parameters:
Maximum number of word matches (-t) — an upper limit of word matches between a read and other reads. Increasing the value would result in more accuracy, however this could slow down the program. The specified value should be more than 0.
Band expansion size (-a) — a number of bases to expand a band of diagonals for an overlapping alignment between two sequence reads. The specified value should be more than 10.
Max gap length in any overlap (-f) — reject overlaps with a gap longer than the specified value. A small value may cause the program to remove true overlaps and to produce incorrect results. This option may be used by the user to split reads from alternative splicing forms into separate contigs. The specified value should be more than 1.
Assembly reverse reads (-r) — consider reads in reverse orientation for assembly. The default value is “checked”.