SPAdes – St. Petersburg genome assembler. Click this link to open SPAdes homepage. SPAdes is embedded as an external tool into UGENE.
SPAdes tool is available on macOS and Linux operating systems only.
Open Tools ‣ NGS data analysis.
Select the Genome de novo assembly item to use the SPAdes.
The Assemble Genomes dialog will appear.
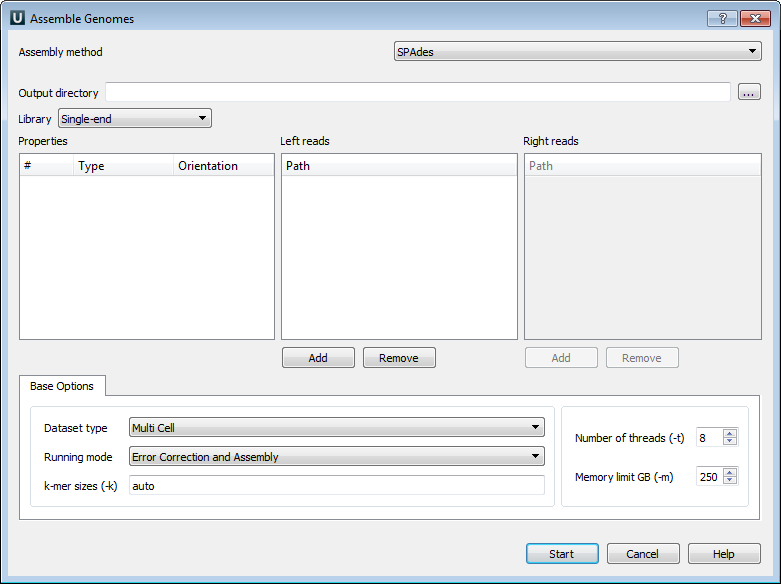
The following parameters are available:
Output directory - SPAdes stores all output files in output directory, which is set by the user.
Library - to run SPAdes choose one of the following libraries:
- Single-end
- Paired-end
- Paired-end (Interplaced)
- Paired-end (Unpaired files)
- Sanger
- PacBio
Left reads - file(s) with left reads.
Right reads - file(s) with right reads.
For each dataset in the paired-end libraries you can change type and orientation.
Datasest type - dataset type.
Running mode - running mode.
k-mer sizes (-k) - k-mer sizes.
Number of threads (-t) - number of threads.
Memory limit GB (-m) - memory limit.