The workflow sample, described below, call variants for an input assembly and a reference sequence using SAMtools mpileup and bcftool. Predict effects of the variants using SnpEff.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Variant Calling and Effect Prediction" can be found in the "NGS" section of the Workflow Designer samples.
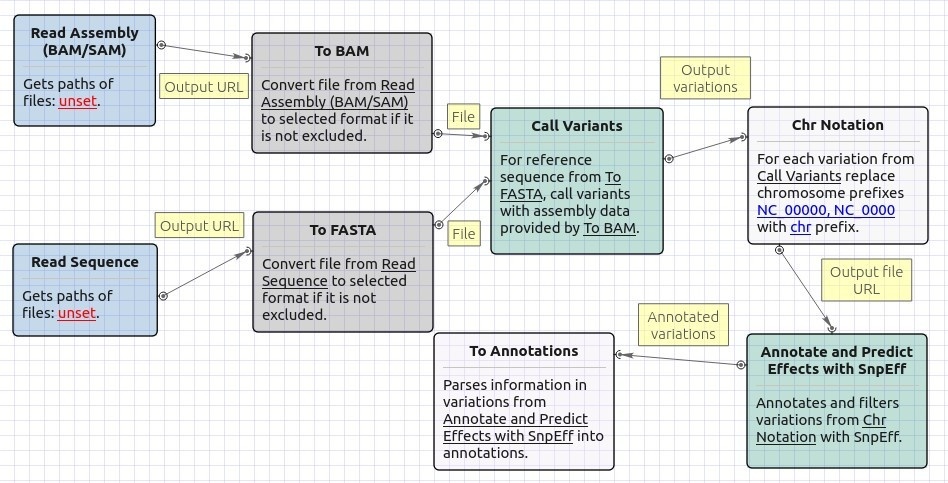
Workflow Image
The opened workflow looks as follows:
Workflow Wizard
The wizard has 7 pages.
Input reference sequence and assembly On this page, input files must be set.
SAMtools mpileup parameters: The SAMtoolsmpileup parameters can be changed here.
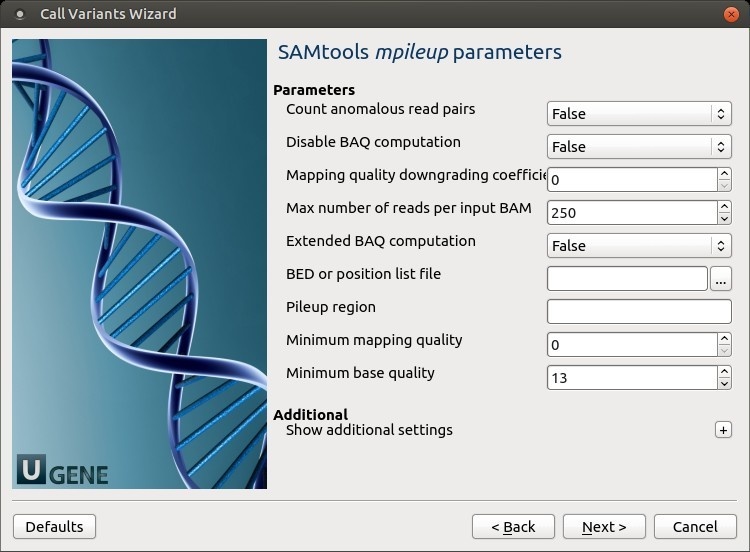
The following parameters are available:
Count anomalous read pairs Do not skip anomalous read pairs in variant calling(mpileup)(-A).
Disable BAQ computation Disable probabilistic realignment for the computation of base alignment quality (BAQ). BAQ is the Phred-scaled probability of a read base being misaligned. Applying this option greatly helps to reduce false SNPs caused by misalignments. (mpileup)(-B).
Mapping quality downgrading coefficient Coefficient for downgrading mapping quality for reads containing excessive mismatches. Given a read with a phred-scaled mapping quality q of being generated from the mapped position, the new mapping quality is about sqrt((INT-q)/INT)*INT. A zero value disables this functionality; if enabled, the recommended value for BWA is 50 (mpileup)(-C).
Max number of reads per input BAM At a position, read maximally the number of reads per input BAM (mpileup)(-d).
Extended BAQ computation Extended BAQ computation. This option helps sensitivity especially for MNPs, but may hurt specificity a little bit (mpileup)(-E).
BED or position list file BED or position list file containing a list of regions or sites where pileup or BCF should be generated (mpileup)(-l).
Pileup region Only generate pileup in region STR (mpileup)(-r).
Minimum mapping quality Minimum mapping quality for an alignment to be used (mpileup)(-q).
Minimum base quality Minimum base quality for a base to be considered (mpileup)(-Q).
Illumina-1.3+ encoding Assume the quality is in the Illumina 1.3+ encoding (mpileup)(-6).
Gap extension error Phred-scaled gap extension sequencing error probability. Reducing INT leads to longer indels (mpileup)(-e).
Homopolymer errors coefficient Coefficient for modeling homopolymer errors. Given an l-long homopolymer run, the sequencing error of an indel of size s is modeled as INT*s/l (mpileup)(-h).
No INDELs Do not perform INDEL calling (mpileup)(-I).
Max INDEL depth Skip INDEL calling if the average per-sample depth is above INT (mpileup)(-L).
Gap open error Phred-scaled gap open sequencing error probability. Reducing INT leads to more indel calls (mpileup)(-o).
List of platforms for indels Comma delimited list of platforms (determined by @RG-PL) from which indel candidates are obtained.It is recommended to collect indel candidates from sequencing technologies that have low indel error rate such as ILLUMINA (mpileup)(-P).
SAMtools bcftools view parameters: The SAMtoolsbcftools parameters can be changed here.
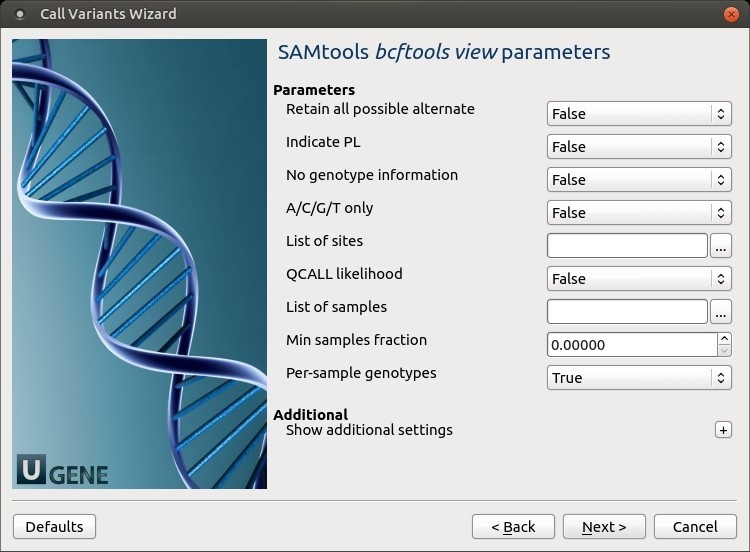
The following parameters are available:
Retain all possible alternate Retain all possible alternate alleles at variant sites. By default, the view command discards unlikely alleles.
Indicate PL Indicate PL is generated by r921 or before (ordering is different).
No genotype information Suppress all individual genotype information.
A/C/G/T only Skip sites where the REF field is not A/C/G/T.
List of sites List of sites at which information are outputted.
QCALL likelihood Output the QCALL likelihood format.
List of samples List of samples to use. The first column in the input gives the sample names and the second gives the ploidy, which can only be 1 or 2. When the 2nd column is absent, the sample ploidy is assumed to be 2. In the output, the ordering of samples will be identical to the one in FILE. Min samples fraction Skip loci where the fraction of samples covered by reads is below FLOAT.
Per-sample genotypes Call per-sample genotypes at variant sites.
INDEL-to-SNP Ratio Ratio of INDEL-to-SNP mutation rate.
Max p(ref|D) A site is considered to be a variant if P(ref|D).
Prior allele frequency spectrum If STR can be full, cond2, flat or the file consisting of error output from a previous variant calling run (bcf view)(-P).
Mutation rate Scaled mutation rate for variant calling (bcf view)(-t).
Pair/trio calling Enable pair/trio calling. For trio calling, option -s is usually needed to be applied to configure the trio members and their ordering. In the file supplied to the option -s, the first sample must be the child, the second the father and the third the mother. The valid values of STR are “pair”, “trioauto”, “trioxd” and “trioxs”, where “pair” calls differences between two input samples, and “trioxd” (“trioxs”) specifies that the input is from the X chromosome non-PAR regions and the child is a female (male). N group-1 samples Number of group-1 samples. This option is used for dividing the samples into two groups for contrast SNP calling or association test. When this option is in use, the following VCF INFO will be outputted: PC2, PCHI2 and QCHI2. N permutations Number of permutations for association test (effective only with -1).
Max P(chi^2) Only perform permutations for P(chi^2). SAMTolls vcfutils varFilter parameters: The next page allows one to configure SAMtools vcfutils parameters.
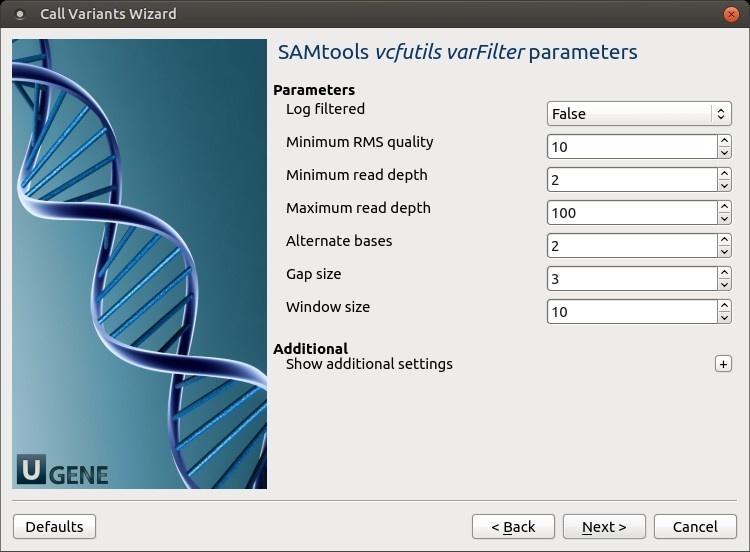
The following parameters are available:
Log filtered
Print filtered variants into the log (varFilter) (-p).
Minimum RMS quality
Minimum RMS mapping quality for SNPs (varFilter) (-Q).
Minimum read depth
Minimum read depth (varFilter) (-d).
Maximum read depth
Maximum read depth (varFilter) (-D).
Alternate bases
Minimum number of alternate bases (varFilter) (-a).
Gap size
SNP within INT bp around a gap to be filtered (varFilter) (-w).
Window size
Window size for filtering adjacent gaps (varFilter) (-W).
Strand bias
Minimum P-value for strand bias (given PV4) (varFilter) (-1).
BaseQ bias
Minimum P-value for baseQ bias (varFilter) (-2).
MapQ bias
Minimum P-value for mapQ bias (varFilter) (-3).
End distance bias
Minimum P-value for end distance bias (varFilter) (-4).
HWE
Minimum P-value for HWE (plus F<0) (varFilter) (-e).
Change chromosome notation for variations: The next page allows change chromosome notation for variations.
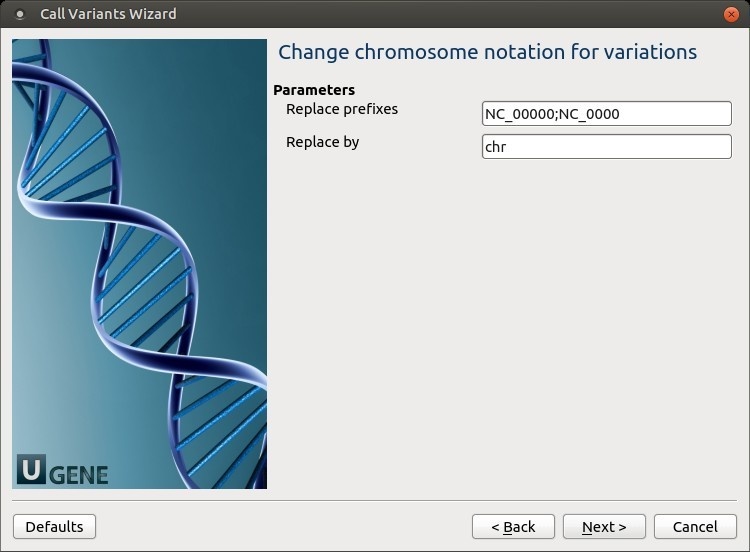
The following parameters are available:
Replace prefixes
Input the list of chromosome prefixes that you would like to replace, for example, "NC_000". Separate different prefixes by semicolons. Replace by
Input the prefix that should be set instead, for example, "chr".
SnpEff parameters: The next page allows one to configure SnpEff parameters.
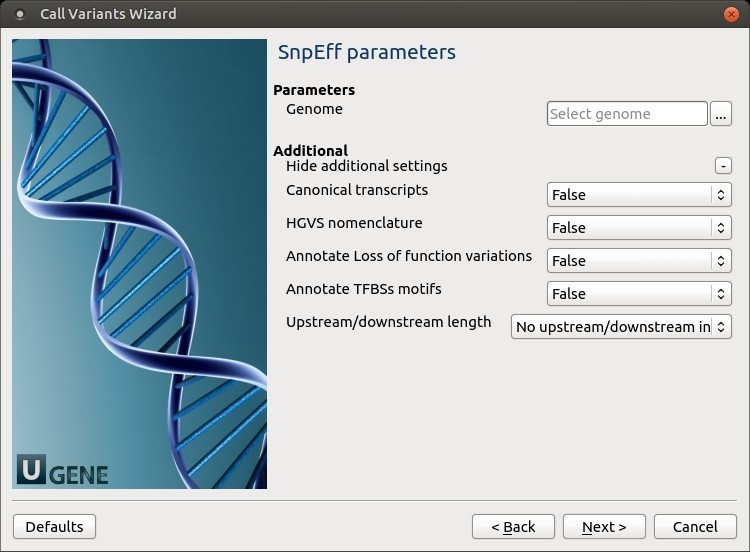
The following parameters are available:
Genome
Select the target genome. Genome data will be downloaded if it is not found.
Canonical transcripts
Use only canonical transcripts
HGVS nomenclature
Annotate using HGVS nomenclature
Annotate Loss of function variations
Annotate Loss of function variations (LOF) and Nonsense mediated decay (NMD)
Annotate TFBSs motifs
Annotate transcription factor binding site motifs (only available for latest GRCh37)
Upstream/downstream length
Upstream and downstream interval size. Eliminate any upstream and downstream effect by using 0 length
Output files Page: On this page, output files can be selected: