Download and install the UGENE FULL or NGS package to use this pipeline.
Use this workflow sample to process raw RNA-seq next-generation sequencing (NGS) data from the Illumina platform. The processing includes:
- Filtration:
- Filtering of the NGS short reads by the CASAVA 1.8 header;
- Trimming of the short reads by quality;
- [Optionally] Mapping:
- Mapping of the short reads to the specified reference sequence (the TopHat tool is used in the sample);
The result output of the workflow contains the filtered and merged FASTQ files. In case the TopHat mapping has been done, the result also contains the TopHat output files: the accepted hits BAM file and tracks of junctions, insertions and deletions in BED format. Other intermediate data files are also output by the workflow.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
What's Next?
Workflow Sample Location
The workflow sample "Raw DNA-Seq processing" can be found in the "NGS" section of the Workflow Designer samples.
Workflow Image
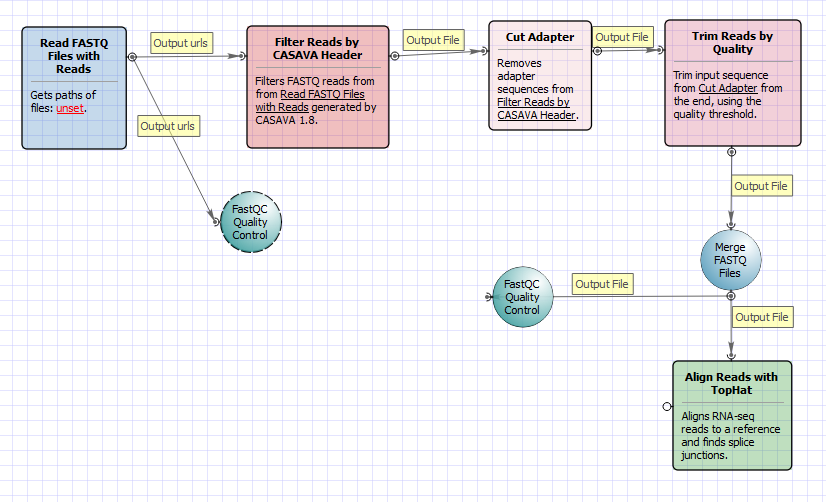
There are four versions of the workflow available. The workflow with mapping for single-end reads looks as follows:
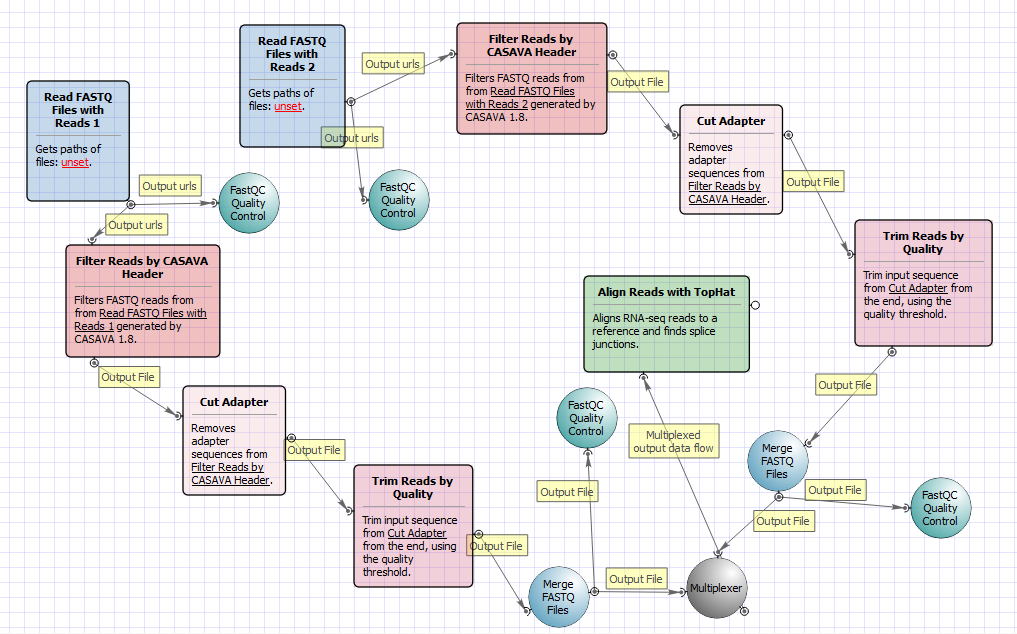
The workflow with mapping for paired-end short appearance is the following:
Workflow Wizard
The wizard for single-end reads has 5 page.
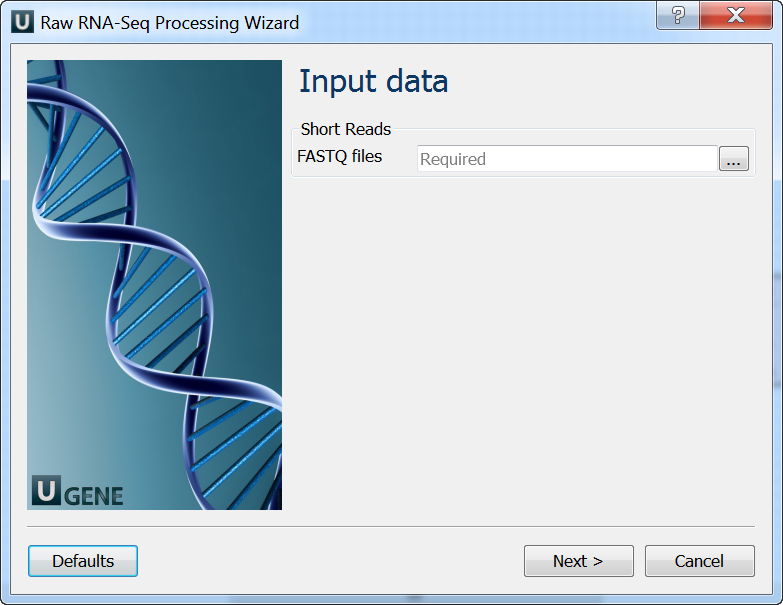
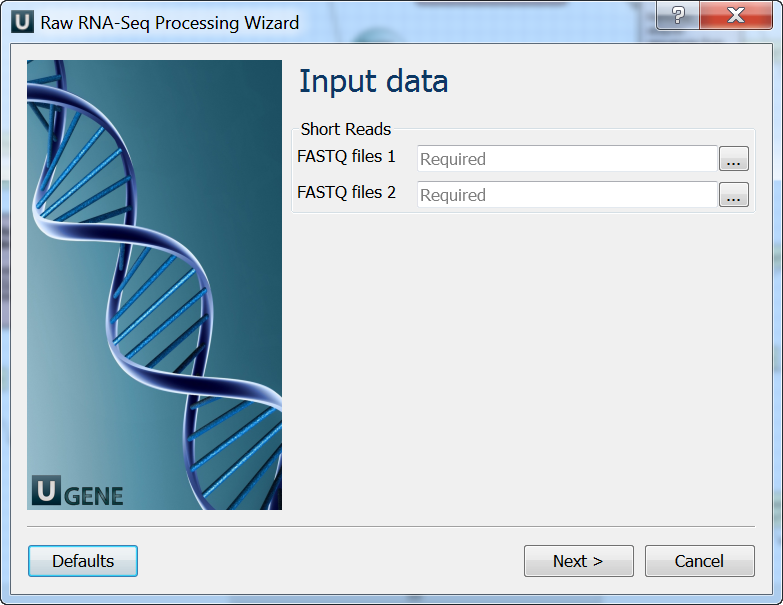
Input data: On this page you must input FASTQ file(s).
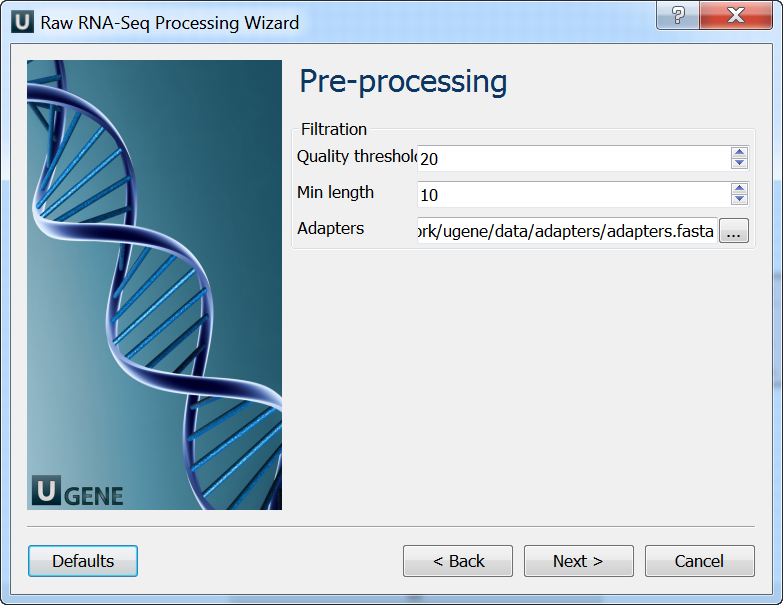
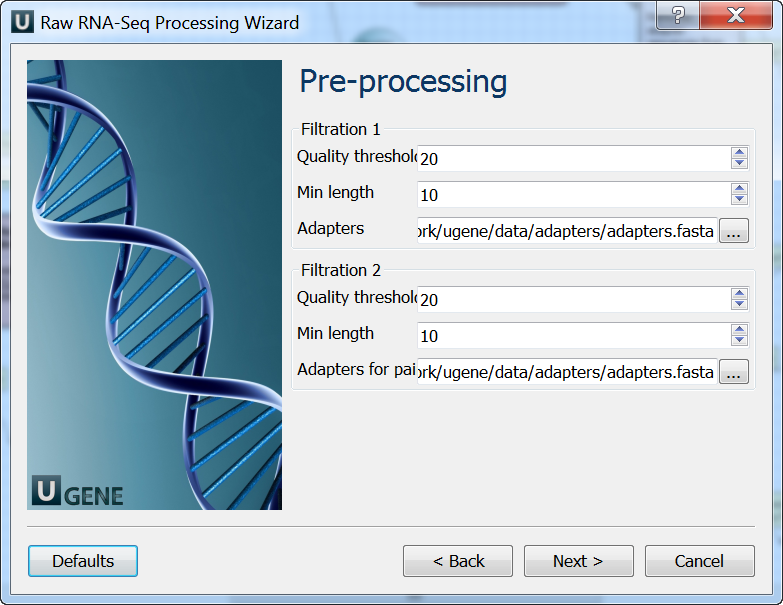
Pre-processing: On this page you can modify filtration parameters.
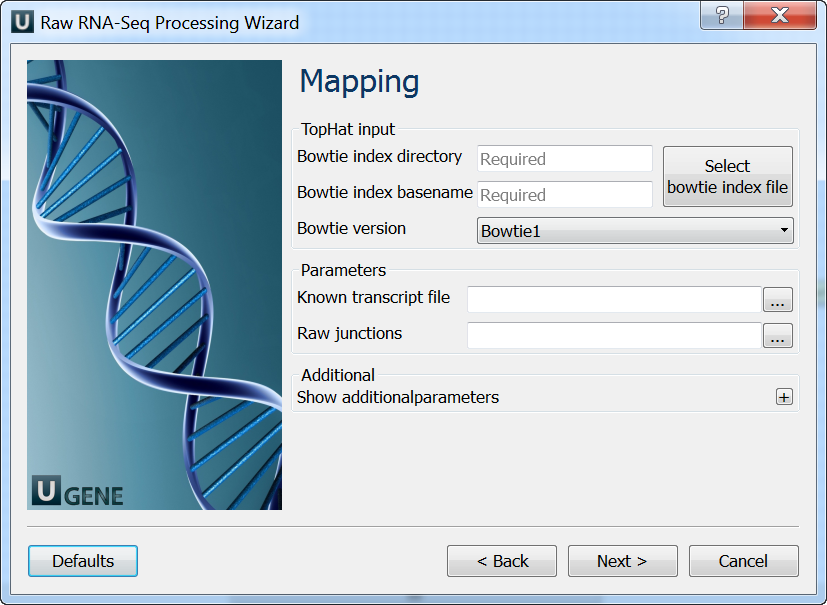
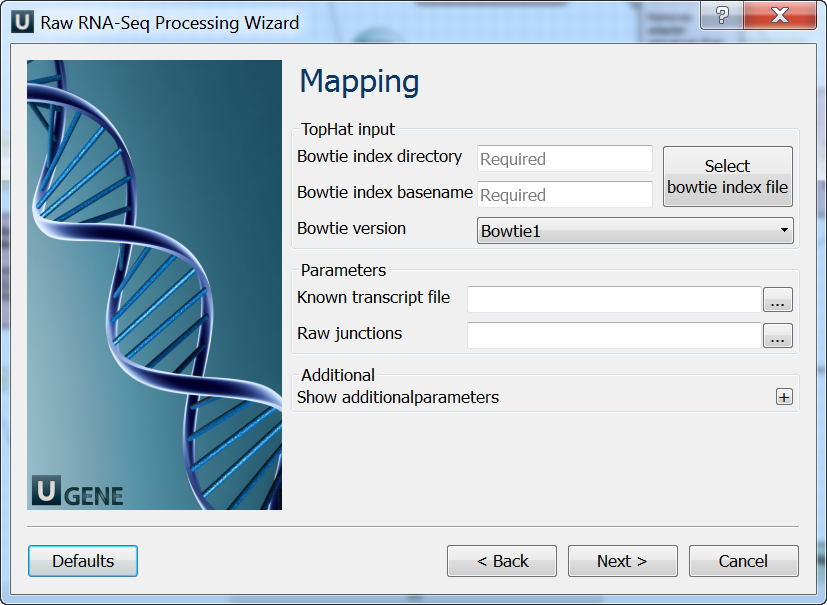
Mapping: On this page you must input reference and optionally modify advanced parameters.
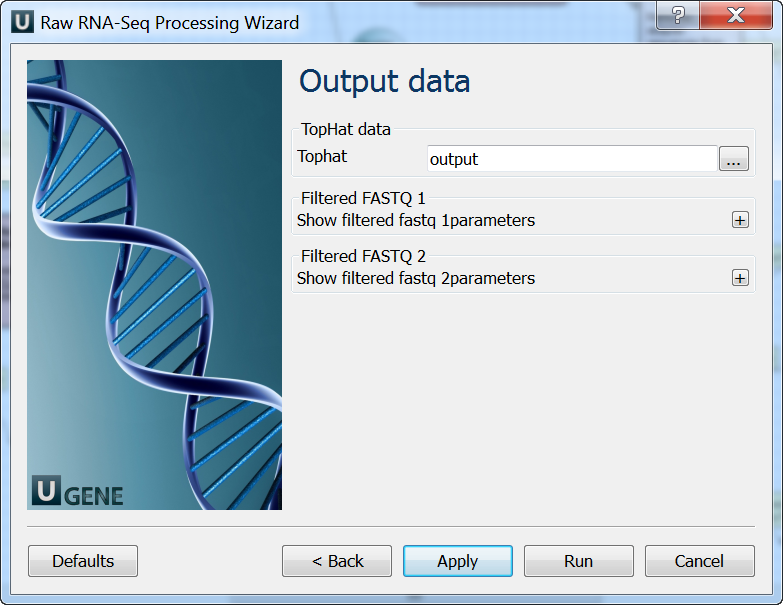
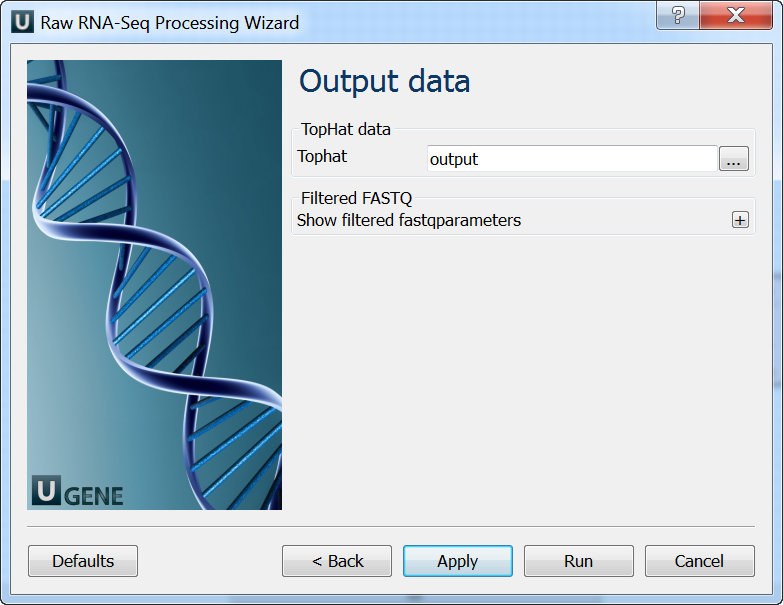
Output data: On this page you must input output parameters.
The wizard for paired-end reads has 5 page.
Input data: On this page you must input FASTQ file(s).
Pre-processing: On this page you can modify filtration parameters.
Mapping: On this page you must input reference and optionally modify advanced parameters.
Output data: On this page you must input output parameters.