When you first open UGENE, you'll see the empty UGENE window with main settings. With a help of this settings you can run the many algorithms, configure different settings and use the help menu if you need it.
After that you open the UGENE main window you can select and analyze different files such as sequences, multiple sequence alignments, short reads assemblies and etc. To open a file click to the File-->Open or click to the Open icon and choose the file:
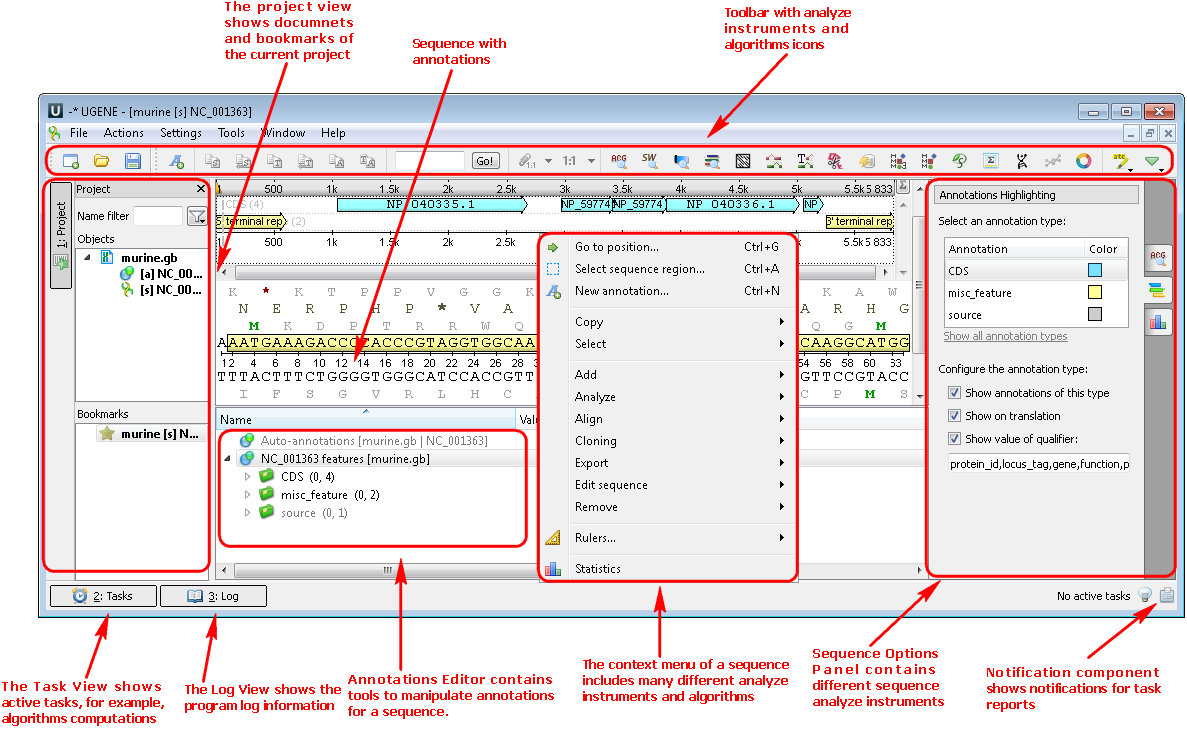
1. If you choose a file with a nucleic or protein sequence you can see the following picture where there are the project view with your document, sequence view with your sequence and its annotations, options panel with many available options. You can also use algorithms and instruments from the context menu:
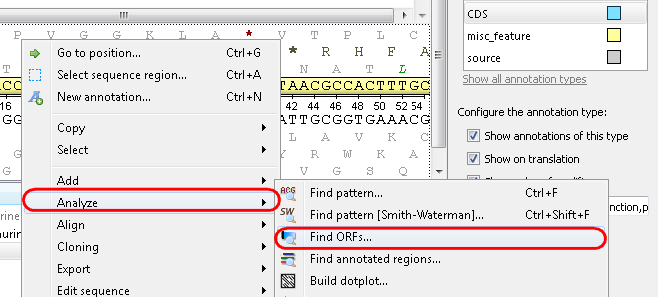
Example 1: For example, you want to find open reading frames (ORFs) in your sequence. For this select the Analyze-->Find ORFs item from the context menu:
or by the toolbar Show ORFs icon:
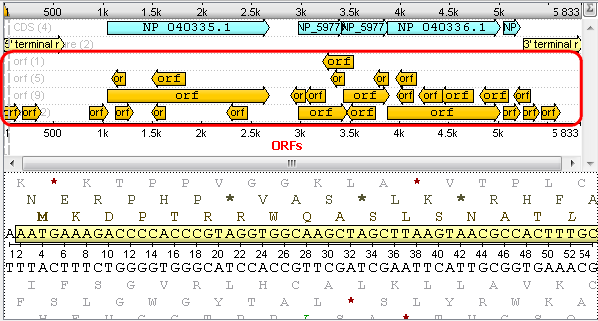
After that you will see your sequence with ORFs:
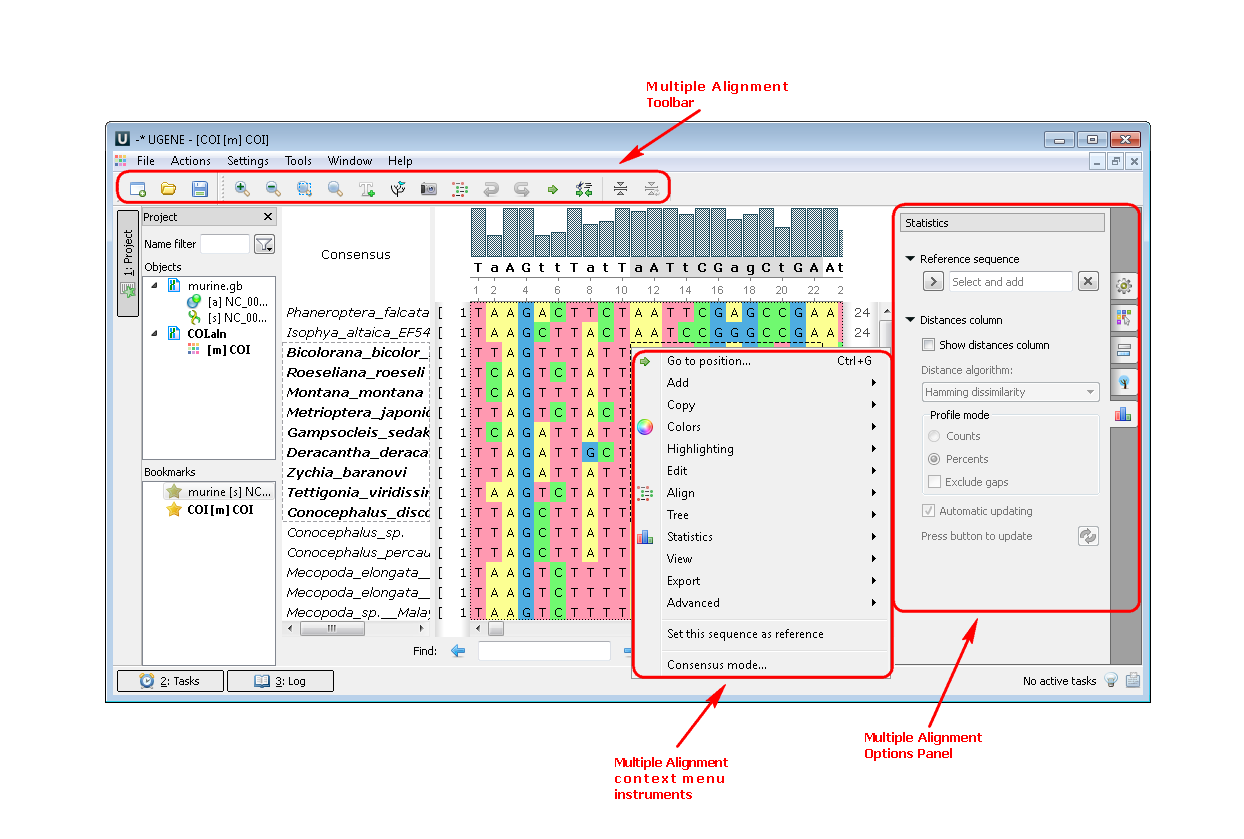
2. If you open a file with a multiple sequence alignment you can see the following picture where there are Options Panel, Toolbar and context menu with many instruments and algorithms:
Example 2: For example, you may want to build a tree from your alignment. You can do this by three different ways:
a. From the toolbar:

b. From the context menu:
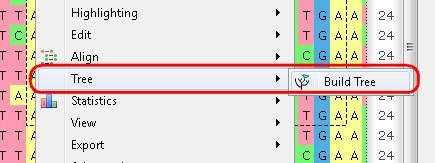
c. From the Options Panel:
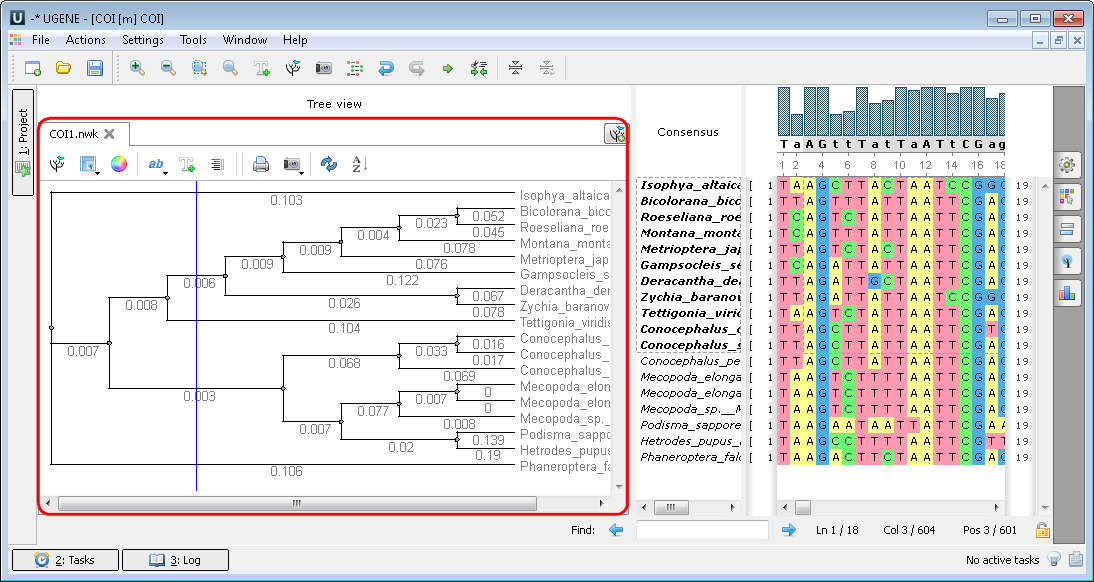
When you choose the building tree parameters in the dialogs the tree appears:
3. If you click a file a short reads assembly, like BAM or SAM format you see the following picture:
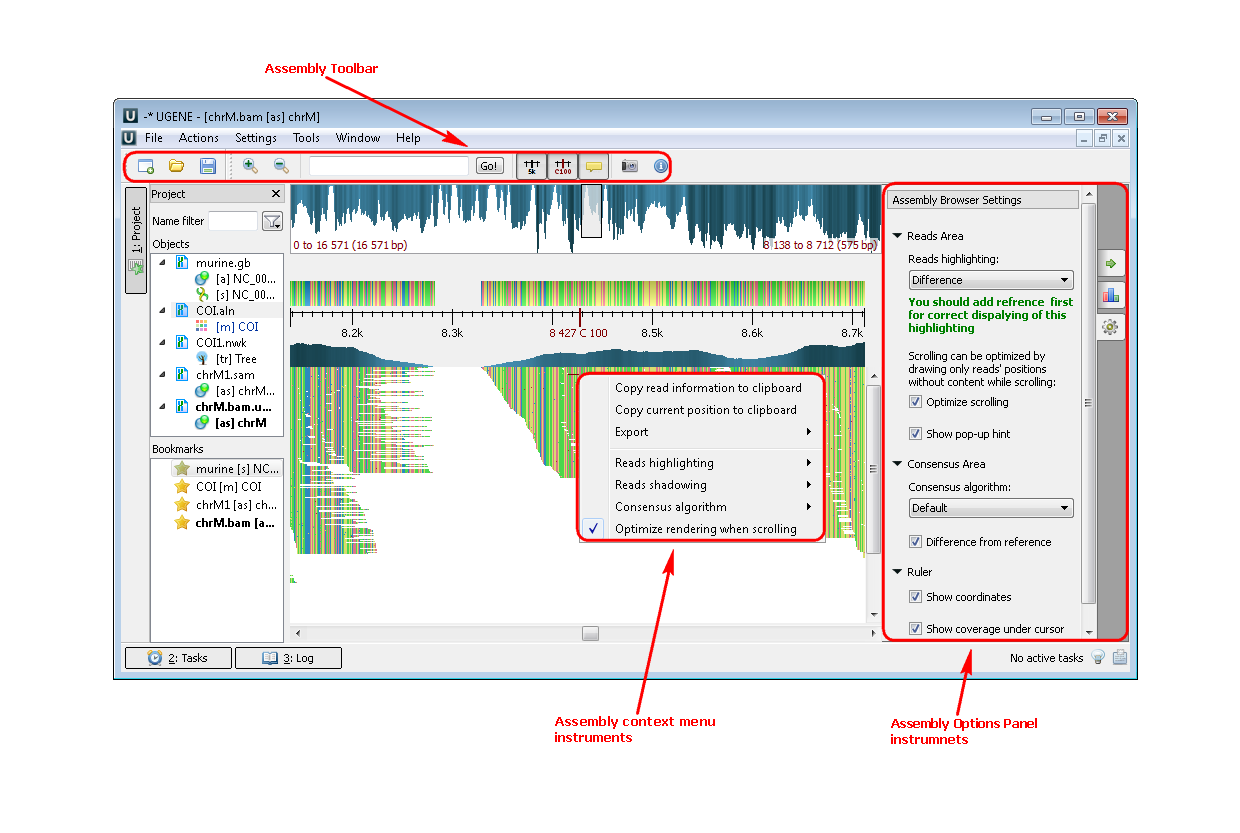
Example 3: For example, you want to highlight the strand direction reads. You can do this by two ways: by the context menu or by the Options Panel menu.
.png)
These were the basic functions provided for you by UGENE. Read more about advanced options in the next chapter.