When you open UGENE for the first time, there is an empty UGENE window with the main menu bar.
Using these menus you can run many algorithms, configure different settings and get help.
- Creating, editing and annotating of nucleotide and protein sequences
- Creating, editing, aligning of multiple sequence alignments
- Assembling, browsing and analyzing of short reads assemblies
- 3D structure viewer
- Building and visualizing of phylogenetic trees
- Running dozens of algorithms for analyzing
- Importing and exporting data into different bio-formats
- Combining algorithms into custom workflows with UGENE Workflow Designer
You can select and analyze different files such as sequences, multiple sequence alignments, short reads assemblies and etc. To open a file click on the File-->Open or click on the Open icon and choose the file:
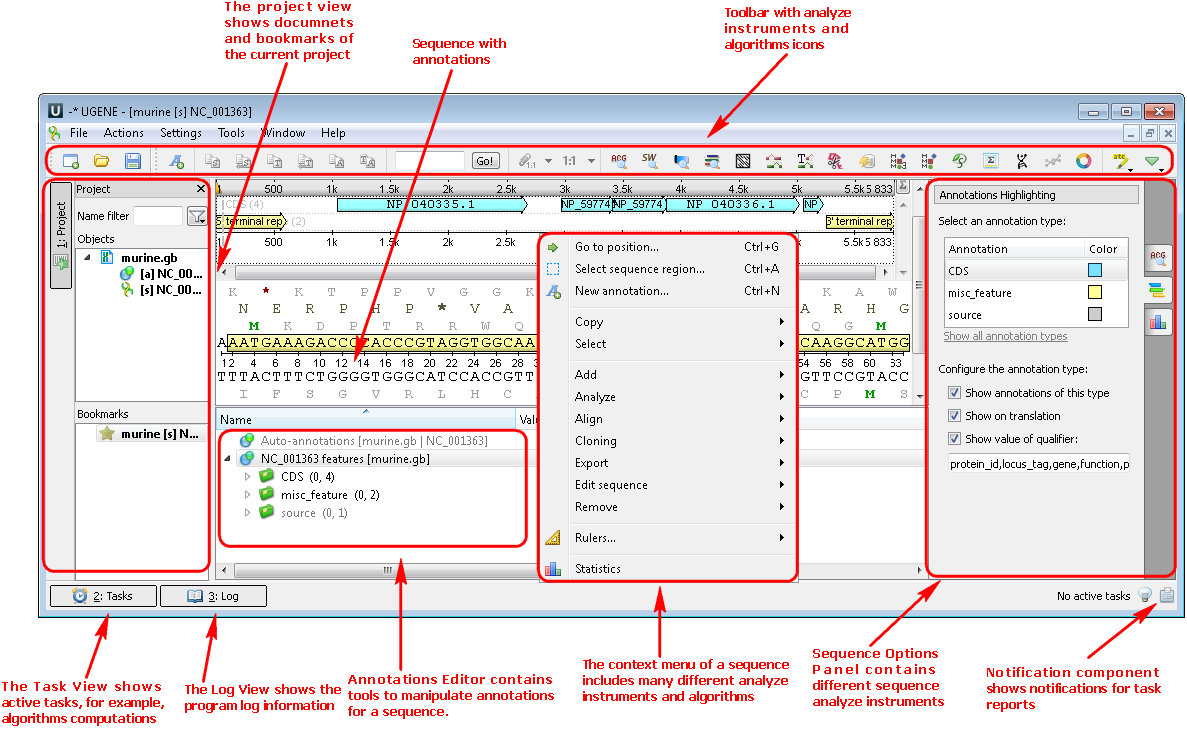
1. Nucleotide or protein sequences (Sequence View). If you open a sequence the following views are available: the project view with your documents, the sequence view with your sequence and its annotations, options panel with additional info and algorithms. You can also use algorithms and instruments from the context menu or the toolbar:
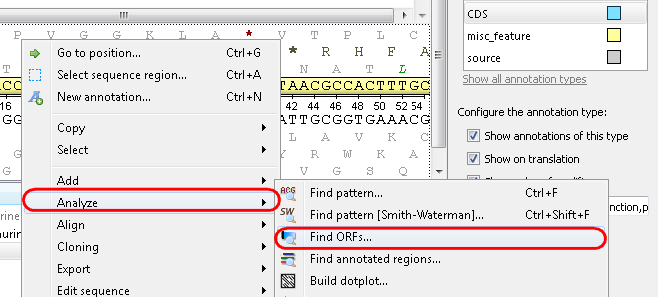
Example 1: Finding open reading frames (ORFs) in your sequence. Select the Analyze-->Find ORFs item from the context menu:
Or click on the Show ORFs button on the toolbar :
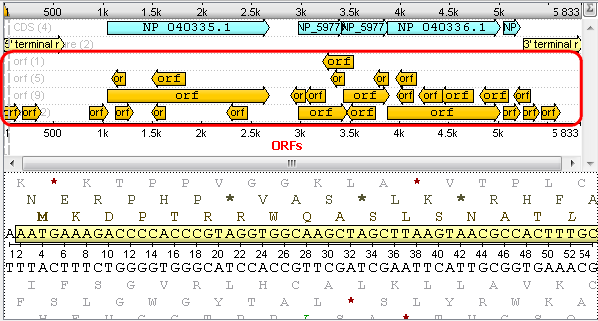
After that you will see your sequence with ORFs:
2. Multiple sequence alignment (MSA Editor). If you open a multiple sequence alignment the following views are available: the project view with your documents, the MSA Editor with your alignment, options panel with additional info and algorithms. You can also use algorithms and instruments from the context menu or the toolbar:
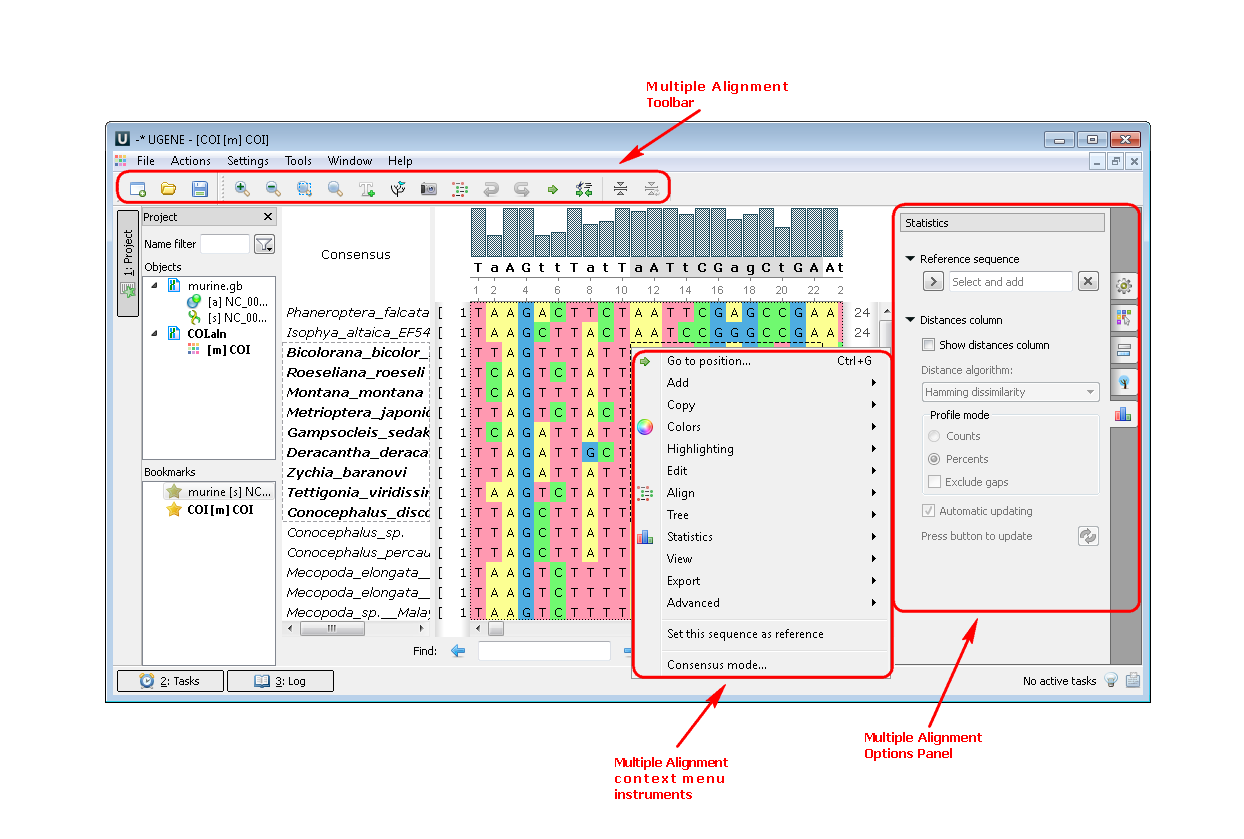
Example 2:Build a tree from your alignment. You can do this by three different ways:
a. From the toolbar:

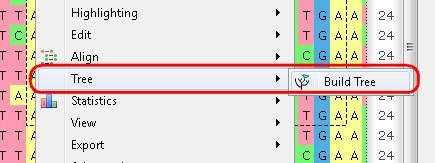
b. From the context menu:
c. From the Options Panel:
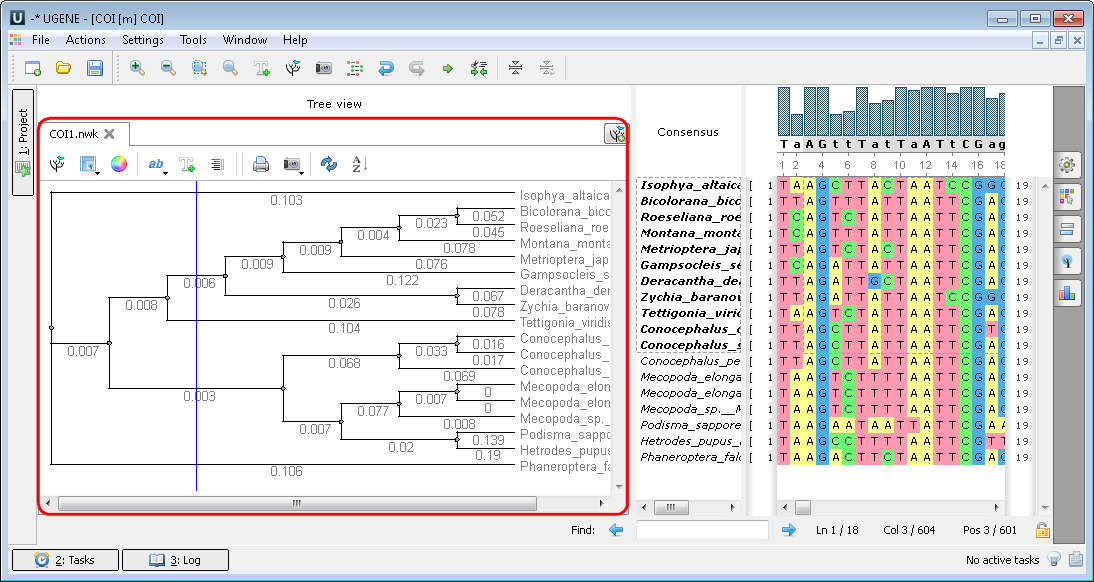
After calculation the tree appears in the MSA Editor in a separate window:
3. SAM/BAM files (Assembly Browser). If you open a file with a short reads assembly, like BAM or SAM files you see the following picture:
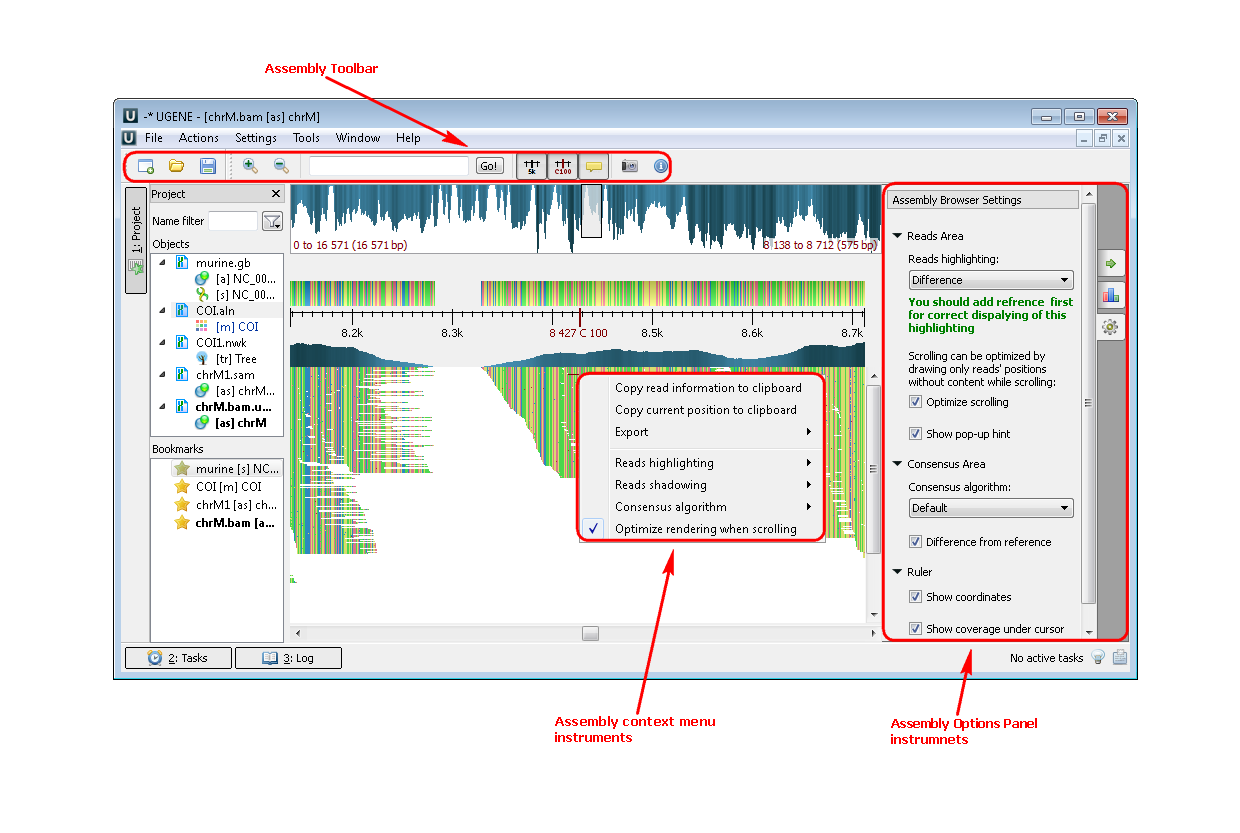
Example 3: Highlighting the strand of reads. You can do this using the context menu or the Options Panel.
.png)