To build BWA-SW index select the Tools ‣ NGS data analysis ‣ Build index for reads mapping item in the main menu. The Build Index dialog will appears. Set the Align short reads method parameter to BWA-MEM.
The dialog looks as follows:
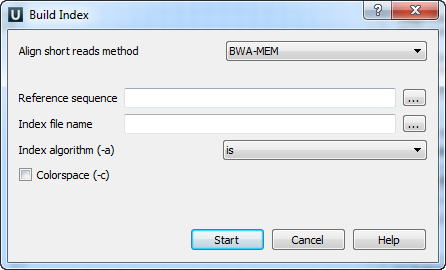
There are the following parameters:
Reference sequence — DNA sequence to which short reads would be aligned to. This parameter is required.
Index file name — file to save index to. This parameter is required.
Index algorithm (-a) — Algorithm for constructing BWA index. Available options are:
It implements three different algorithms
- is — designed for short reads up to ~200bp with low error rate (<3%). It does gapped global alignment w.r.t. reads, supports paired-end reads, and is one of the fastest short read alignment algorithms to date while also visiting suboptimal hits.
- bwtsw — is designed for long reads with more errors. It performs heuristic Smith-Waterman-like alignment to find high-scoring local hits. Algorithm implemented in BWA-SW. On low-error short queries, BWA-SW. is slower and less accurate than the is algorithm, but on long reads, it is better.
- div — does not work for long genomes.
The value "autodetect" by default means that the index (one of three) is chosen automatically depending on the length of the sequence.