The 'mfold' software is a tool for nucleic acid folding and hybridization prediction. It was developed by M. Zuker in the late 1980s for RNA folding and improved for DNA folding in 1996. mfold uses nearest neighbor energy rules.
Tool home page: mfold.org. Similar results to those produced by UGENE can be obtained using the mfold web server (DNA prediction, RNA prediction) or using the OligoAnalyzer tool. For complete documentation on the tool, see a) on the website, b) in the article, c) in the source code in the corresponding folder.
UGENE uses Ghostscript to convert PS files to PNG and PDF.
ToC
Open the dialog
mfold only works with DNA or RNA sequences. You can trigger the dialog from
by selecting the appropriate Sequence View Global action 
or through the Main Menu→Actions→Analyze 
or through the Sequence Context Menu→Analyze 
This is what the mfold dialog looks like
Input parameters
The tool is passed explicit and implicit parameters. Explicit parameters are set in the dialog, implicit parameters are taken from the sequence context.
Algorithm settings
For complete settings details, see the documentation on the mfold website or documentation in source.
This is the Settings section of the mfold settings tab.
| Parameter | Unit | Default | Limits | Tool argument name | Description |
|---|---|---|---|---|---|
| Temperature | °C | 37 | [0,100] | T | The folding temperature. For RNA this is always the default value. For DNA it is taken from the dialog. |
| Ionic conditions | M | Na=1 | [0,1.5] | NA_CONC | Ionic conditions are used to enter total monovalent (Na) and divalent (Mg) ions concentrations. For RNA this is always the default values. For DNA it is taken from the dialog. |
| Mg=0 | MG_CONC | ||||
| Percent suboptimality | % | 5 | [1,100] | P | The suboptimality percentage controls the free energy increment δδG for computing suboptimal foldings. Only foldings with a free energy ≤ΔG+δδG will be computed, where ΔG is the predicted minimum free energy. Normally, δδG=(P/100)|ΔG|, but it is rounded up to 1 kcal/mol or down to 12 kcal/mol if it is outside this range. |
| Max num of foldings | 50 | [1,100] | MAX | This is the maximum number of foldings that mfold will compute. It is better to limit the number of foldings by careful selection of the P and W parameters. | |
| Window | 0 (len≤29) 1 (30≤len≤49) 2 (50≤len≤119) 3 (120≤len≤199) 5 (200≤len≤299) 7 (300≤len≤399) 8 (400≤len≤499) 10 (500≤len≤599) 11 (600≤len≤699) 12 (700≤len≤799) 15 (800≤len≤1199) 20 (1200≤len≤1999) 25 (2000≤len) | [0,50] | W | The window parameter, W, controls the number of foldings that are computed. It may be thought of as a distance parameter. The distance between 2 base pairs, ri rj and ri' rj' may be defined as max{|i-i′|, |j-j′|}. If k-1 foldings have already been predicted by mfold, the kth folding must have at least W base pairs that are at least a distance W from any of the base pairs in the first k-1 foldings. A new folding is not added to the output list unless this criterion is fulfilled. As W increases, the number of predicted foldings decreases. A smaller value of this parameter will usually result in more computed foldings that may be quite similar to one another. A larger value will result in fewer foldings that are very different from one another. If the parameter is set to the default value, then in the calculations the real value of W will be set according to the described algorithm, based on the length of the selected region of the input sequence (see the Default column). | |
| Max base pair distance | ∞ | [1,∞] | MAXBP | If the maximum distance between paired bases parameter, MAXBP, is specified, then any base pair, ri rj, in a folding must satisfy Thus small values of MAXBP ensure that only short range base pairs will be predicted. For example, in a sequence of 1000 nucleotides, setting MAXBP to 50 will force mfold to compute foldings involving only short range base pairs. The default is MAXBP=∞, which means no constraint. |
Display settings
Corresponds to the Extended settings section on the main tab.
| Parameter | Unit | Default | Limits | Tool argument name | Description |
|---|---|---|---|---|---|
| Base numbering frequency | 10 (len≤50) 20 (51≤len≤300) 50 (301≤len) | [0,1000] | LAB_FR | Each image marks the number of the base (nucleotide) starting from the beginning 5'. The frequency with which this number will be displayed on an image depends on this parameter: Compare images with the same algorithm settings to see the difference. | |
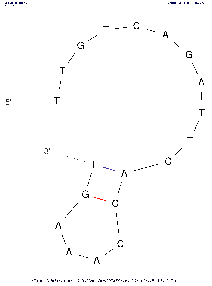
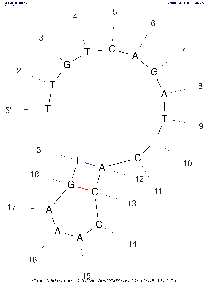
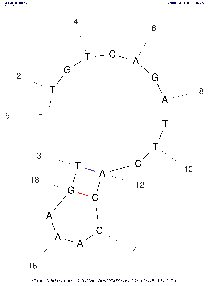
Tool comparison