This section contains detailed description of all workflow elements presented in the Workflow Designer.
For each element you can find:
- Description of the parameters used in the GUI
- Corresponding parameters names used in a workflow file
- Information about input and output ports
The type of a parameter can be one of the following:
string
A string.
numeric
A number.
boolean
A boolean data type. Available values are: true / false, 0 / 1 and yes / no.
A port’s slot type can be one of the following:
sequence
Biological sequence
msa
Multiple sequence alignment
text
A text
annotation-table
Table of annotations
annotation-table-list
A list of different tables of annotations
ebwt-index
Bowtie index
hmm2-profile
A HMM profile of HMMER2 package
fmatrix
Frequency matrix
wmatrix
Weight matrix
sitecon-model
SITECON model
assembly
Assembly
variation
Variation track
To search an element use the name filter or press the Ctrl+F shortcut that moves you to the name filter also:
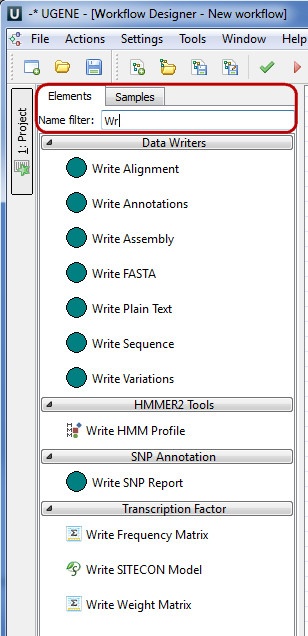
- Data Readers
- Data Writers
- Data Flow
- Basic Analysis
- Amino Translations Element
- Annotate with UQL Element
- CD-Search Element
- Collocation Search Element
- Export PHRED Qualities Element
- Fetch Sequences by ID From Annotation Element
- Filter Annotation by Name Element
- Filter Annotations by Qualifier
- Find Correct Primer Pairs Element
- Find Pattern Element
- Find Repeats Element
- Gene-by-gene approach report
- Get Sequences by Annotations Element
- Group Primer Pairs Element
- Import PHRED Qualities Element
- Intersect Annotations Element
- Local BLAST Search Element
- Local BLAST+ Search Element
- Merge Annotations Element
- ORF Marker Element
- Remote BLAST Element
- Sequence Quality Trimmer Element
- Smith-Waterman Search Element
- Data Converters
- DNA Assembly
- HMMER2 Tools
- HMMER3 Tools
- Multiple Sequence Alignment
- Align Profile to Profile with MUSCLE Element
- Align with ClustalO Element
- Align with ClustalW Element
- Align with Kalign Element
- Align with MAFFT Element
- Align with MUSCLE Element
- Align with T-Coffee Element
- Extract Consensus from Alignment as Sequence
- Extract Consensus from Alignment as Text
- In Silico PCR Element
- Join Sequences into Alignment Element
- Map to Reference Element
- Split Alignment into Sequences Element
- NGS: Basic Functions
- CASAVA FASTQ Filter Element
- Cut Adapter Element
- Extract Consensus from Assembly Element
- Extract Coverage from Assembly Element
- FASTQ Merger Element
- FASTQ Quality Trimmer Element
- FastQC Quality Control Element
- Filter BAM/SAM Files Element
- Genome Coverage Element
- Improve Reads with Trimmomatic Element
- Merge BAM Files Element
- Remove Duplicates in BAM Files Element
- Slopbed Element
- Sort BAM Files Element
- NGS: ChIP-Seq Analysis
- NGS: Map/Assemble Reads
- NGS: Metagenomics Classification
- Build CLARK Database
- Build DIAMOND Database
- Build Kraken Database
- Classification Report Element
- Classify Sequences with CLARK
- Classify Sequences with DIAMOND
- Classify Sequences with Kraken
- Classify Sequences with MetaPhlAn2
- Ensemble Classification Data
- Filter by Classification
- Improve Classification with WEVOTE
- NGS: RNA-Seq Analysis
- NGS: Variant Analysis
- Transcription Factor
- Build Frequency Matrix Element
- Build SITECON Model Element
- Build Weight Matrix Element
- Convert Frequency Matrix Element
- Read Frequency Matrix Element
- Read SITECON Model Element
- Read Weight Matrix Element
- Search for TFBS with SITECON Element
- Search for TFBS with Weight Matrix Element
- Write Frequency Matrix Element
- Write SITECON Model Element
- Write Weight Matrix Element
- Utils
- Amino Translations Element
- Annotate with UQL Element
- CD-Search Element
- Collocation Search Element
- Export PHRED Qualities Element
- Fetch Sequences by ID From Annotation Element
- Filter Annotation by Name Element
- Filter Annotations by Qualifier
- Find Correct Primer Pairs Element
- Find Pattern Element
- Find Repeats Element
- Gene-by-gene approach report
- Get Sequences by Annotations Element
- Group Primer Pairs Element
- Import PHRED Qualities Element
- Intersect Annotations Element
- Local BLAST Search Element
- Local BLAST+ Search Element
- Merge Annotations Element
- ORF Marker Element
- Remote BLAST Element
- Sequence Quality Trimmer Element
- Smith-Waterman Search Element
- Align Profile to Profile with MUSCLE Element
- Align with ClustalO Element
- Align with ClustalW Element
- Align with Kalign Element
- Align with MAFFT Element
- Align with MUSCLE Element
- Align with T-Coffee Element
- Extract Consensus from Alignment as Sequence
- Extract Consensus from Alignment as Text
- In Silico PCR Element
- Join Sequences into Alignment Element
- Map to Reference Element
- Split Alignment into Sequences Element
- CASAVA FASTQ Filter Element
- Cut Adapter Element
- Extract Consensus from Assembly Element
- Extract Coverage from Assembly Element
- FASTQ Merger Element
- FASTQ Quality Trimmer Element
- FastQC Quality Control Element
- Filter BAM/SAM Files Element
- Genome Coverage Element
- Improve Reads with Trimmomatic Element
- Merge BAM Files Element
- Remove Duplicates in BAM Files Element
- Slopbed Element
- Sort BAM Files Element
- Build CLARK Database
- Build DIAMOND Database
- Build Kraken Database
- Classification Report Element
- Classify Sequences with CLARK
- Classify Sequences with DIAMOND
- Classify Sequences with Kraken
- Classify Sequences with MetaPhlAn2
- Ensemble Classification Data
- Filter by Classification
- Improve Classification with WEVOTE
- Build Frequency Matrix Element
- Build SITECON Model Element
- Build Weight Matrix Element
- Convert Frequency Matrix Element
- Read Frequency Matrix Element
- Read SITECON Model Element
- Read Weight Matrix Element
- Search for TFBS with SITECON Element
- Search for TFBS with Weight Matrix Element
- Write Frequency Matrix Element
- Write SITECON Model Element
- Write Weight Matrix Element